

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Phacomatosis pigmentovascularis (PPV) is a rare syndrome characterized by concurrent nevus flammeus (capillary malformation) and pigmentary nevus, first described in 1947 by Ota et al.[1] In 1985, Hasegawa classified PPV into four types according to the pigmented lesions,[2] with the addition of an additional type by Enjolras and Mulliken,[3] resulting in five different types.[4] Each type is further subtyped depending on the presence of cutaneous only (subtype a) or extracutaneous (subtype b) involvement (Table 1).[5] PPV is often associated with and overlapped with other syndromes. Among them, Sturge-Weber syndrome (SWS) is characterized by facial capillary malformation (port-wine stain), leptomeningeal thickening, and vascular malformation of the choroid of the eye, which is associated with glaucoma.[6] Klippel-Trenaunay syndrome (KTS) is another such syndrome characterized by venous varicosities, cutaneous capillary malformation, and soft tissue overgrowth.[7] In the aforementioned syndromes, there is usually venous involvement. Venous hypertension results from localized primary venous dysplasia, influencing the communicating venous passageways and the compensatory collateral venous channels, and resulting in abnormalities at the venous-capillary level. Cases that involve the arteries are rare.[8] A case of PPV type IIIb resulting in moyamoya disease due to stenosis at the terminal portion of the carotid artery has been reported in the literature,[9] but there has been no reported case of diffuse involvement of multiple systemic arteries.
Table 1.
Types of phacomatosis pigmentovascularis
![]()
We herein report our experience on a patient with PPV type IIb associated with SWS who showed an aneurysm of the posterior communicating artery, renovascular hypertension, intermittent claudication, and severe rhabdomyolysis due to a vascular malformation involving systemic vessels including arteries.
Case Report
The patient was born at full term without perinatal problems. She had nevus flammeus over her face and body, and buphthalmos which was observed since birth. At two years of age, she was diagnosed with glaucoma and underwent an operation. Her growth and development including intelligence were normal. At the age of 9 years, she developed intermittent left sided weakness, blurred vision, and headache, which were mild and self-limited. Six months prior to the current hospital visit at 15 years of age, symptoms were aggravated and the patient complained of headache and intermittent claudication and thus she visited another hospital where magnetic resonance imaging (MRI) of the brain was done. Imaging revealed diffuse leptomeningeal enhancement of the right temporoparietooccipital lobe and peripheral enhancement of the right maxillary sinus, suggesting possible hematoma (Fig. 1). Under the impression of SWS, she was referred to Seoul National University Children’s Hospital for further evaluation.
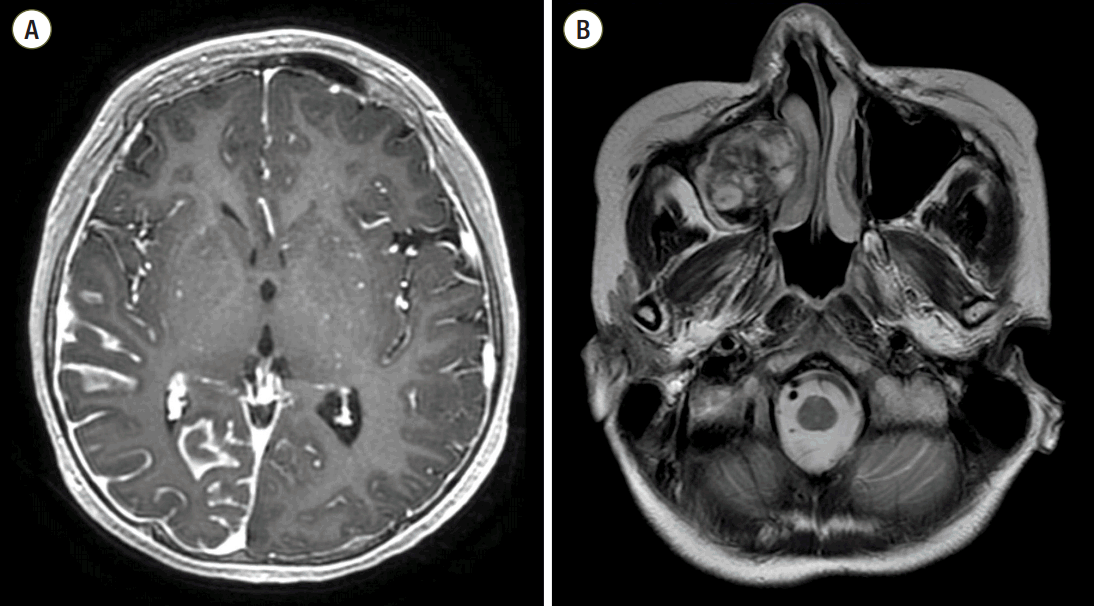
Upon examination, nevus flammeus was distributed extensively over the body and nevus of Ota (oculodermal melanocytosis) was also present (Fig. 2). The patient’s vital signs were stable and within normal range for her age. Laboratory test results were unremarkable. She did not smoke or drink alcohol, and had neither a previous medical history of diabetes mellitus, nor a family history of stroke or headache. An aneurysm of the left posterior communicating artery was evident on brain MRI arteriography (Fig. 3), and she underwent an embolization of the aneurysm with transfemoral cerebral angiography. She underwent hematoma removal of the right maxillary sinus thereafter. The patient’s headache and blurred vision were improved, and she was discharged. Three months after discharge, she visited the outpatient clinic with aggravated headache and intermittent claudication. The blood pressure (BP) was measured at 158/117 mmHg and she was admitted to the general ward.


At admission, the BP was 167/117 mmHg, the heart rate (HR) 92 beats/min, the respiratory rate 22 breaths/min, and the body temperature 36.3. The white blood cell (WBC) counts were elevated to 17,740/mm3, but the C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were normal at 0.48 mg/dL and 17 mm/h respectively. Hypokalemia was detected with a potassium level of 2.6 mmol/L and elevated renin and aldosterone levels measuring 13.47 ng/mL/h and 63.8 ng/dL respectively. Other blood test results were within normal range. Urine catecholamines on 24-hour collection tests were normal. Potassium was supplemented by intravenous (IV) fluids and hypertension treated with oral amlodipine (5 mg bid) as well as continuous infusion of IV nicardipine (0.5-2 μg/kg/min). Computed tomography (CT) imaging of the brain and CT angiography imaging of the arteries of the lower extremities were performed. Focal high-attenuated lesions at the left pons and the right lower pons were suggestive of hemorrhage on the brain CT (Fig. 4), but neurologic deficits were absent and her headache improved, suggesting inactive lesions. There was diffuse narrowing of the arteries of the lower extremities, both renal arteries, inferior vena cava, and both iliac veins on CT angiography without definite evidence of arterial occlusions (Fig. 5A). Bilateral pain of the lower extremities developed the subsequent day, which was controlled with IV pethidine (1 mg/kg/dose). At the time of administration of pethidine, vital signs were within normal range excepting for persistent hypertension (BP 140/90 mmHg). Two hours after administration of pethidine, she developed mental drowsiness, hypotension (BP 72/41 mmHg), desaturation (SpO2 52%), and her light reflex was bilaterally sluggish. The HR dropped to 36 beats/min and cardiopulmonary resuscitation (CPR) was started. Venous blood gas analysis sampled immediately after initiation of CPR showed metabolic acidosis (pH 6.95, pCO2 31.7 mmHg). Blood test results showed hypocalcemia (calcium 3.2 mg/dL), and hyperphosphatemia (phosphorus 26.9 mg/dL). Creatine phosphokinase (CPK) was increased to 185,400 IU/L, lactate dehydrogenase (LDH) to 6,260 IU/L, and myoglobin level to 280.3 ng/dL. There was a return of spontaneous circulation (ROSC) after 19 minutes of CPR and the patient was transferred to the intensive care unit (ICU) for further management.


At the ICU, the pulse of the patient was not palpable and CPR was restarted with ROSC after 8 minutes. Her BP measured 124/54 mmHg and HR 146 beats/min, but spontaneous breathing did not recover and capillary refill time was delayed to 6-7 seconds. Glasgow coma scale (GCS) measured 3t (E1, V1t, M1). Although the light reflex was both prompt, both pupils were dilated to 6 mm (right) and 7 mm (left). The knee jerk was decreased bilaterally to grade 1 and Babinski sign was negative. Follow-up brain CT imaging did not show additional hemorrhagic abnormalities. On the echocardiogram, concentric left ventricular hypertrophy, fractional shortening of 33.7% and ejection fraction of 64.2% was seen. Mechanical ventilator care and fluid management was continued, and continuous renal replacement therapy (CRRT) was started due to rhabdomyolysis and electrolyte imbalance. Dexamethasone (0.3 mg/kg/day IV) was used for the anti-inflammatory effect. Alprostadil (10 μg/day IV), dobutamine (5 μg/kg/min IV continuous infusion), and sodium nitroprusside (0.3-0.5 μg/kg/min IV continuous infusion) were administered to help with perfusion of the narrowed vessels, and enalapril (0.08 mg/kg/day oral) was administered due to renovascular hypertension. To prevent tissue hypoperfusion due to diffuse narrowing of the arteries, the target systolic BP was between 100 and 120 mmHg, and the diastolic BP between 60 and 80 mmHg. Rewarming was started after 24 hours of therapeutic hypothermia. Follow up testing showed exacerbated muscle enzymes and inflammatory markers with CPK of 141,911 IU/L, LDH 9,659 IU/L, myoglobin 1,195,800 ng/dL, CRP 26.15 mg/dL, and procalcitonin 20.13 ng/mL. However, the patient did not have fever and there were no other signs of infection. Her mental status improved gradually, and she was able to obey commands. On doppler ultrasonography of the lower extremity vessels, venous flow of both extremities was intact and there was no evidence of deep vein thrombosis. On the 7th day after admission to the ICU, the patient was successfully extubated and her mentality recovered fully to a GCS of 15. Improvements in blood tests were noted on the 14th day of ICU care and CRRT was discontinued with initiation of intermittent hemodialysis (IHD) and transfer to the general ward.
Self-urination was adequate by the 19th day of hospitalization warranting discontinuation of IHD. IV alprostadil was changed to oral beraprost, and dexamethasone was discontinued because the possibility of ongoing inflammation was low. However, elevated ESR levels to 59 mm/h after discontinuation of dexamethasone lead to initiation of oral deflazacort. Furthermore, evaluation for possible vasculitis by screening anti-nuclear antibodies, rheumatoid factor, anti-double stranded DNA was done and results were negative. Follow-up CT angiography of the lower extremities arteries showed marked improvement of the previously noted diffuse arterial narrowing involving the bilateral common femoral arteries and crural arteries, but there was still remaining stenosis (Fig. 5B). Balloon angioplasty of the right proximal renal artery and right common femoral artery improved their respective residual stenosis to below 30% and 20%, which was evident on the post-angiogram. Hypertension improved and amlodipine was discontinued; systolic BP was maintained between 90 and 110 mmHg and diastolic BP between 45 and 75 mmHg on losartan (1.4 mg/kg/day) alone. The patient’s mental status and blood test results were normal, but the motor power of her lower extremities was decreased to grade IV. She could walk with assistance but could not walk alone. She was transferred to and later discharged from the department of rehabilitation medicine on the 69th day of hospitalization with plans of continuing rehabilitation therapy at a hospital near her house. The patient remains in good condition except for mild weakness of the lower extremities. Her BP is well controlled with medication, and blood test results are normal.
Go to : 
Discussion
PPV is a diagnosis that is made based on clinical morphology. The diagnosis is not too difficult if there is a concurrent capillary malformation (nevus flammeus) and pigmentary nevus. However, in those cases that present with associated systemic manifestations of SWS or KTS, the differential diagnosis is more challenging. The propositus showed widespread nevus flammeus, nevus of Ota, glaucoma, and leptomeningeal enhancement. Consequently, a diagnosis of PPV type IIb with associated SWS is possible in this patient. The current system of diagnosis based on clinical morphology and manifesting signs is indistinct by example of the addition of new types in the event of newly found cases[3,4] and classification as “unclassifiable” in some cases.[10] There are also reports of cases where the clinical features overlap between PPV, SWS, and KTS.[11-13] Therefore, there have been proposals to diagnosis and classification with more comprehensive terms such as “vascular malformation syndrome” rather than SWS or KTS with indefinite boundaries. [14] The overlap may also exist in the pathophysiology as previous reports propose similar mechanisms of nevus flammeus in SWS, KTS and Cobb syndrome as the development of venous hypertension and compensatory collateral channels due to localized primary venous dysplasia.[8] In a recent study, somatic activating mutation in GNAQ gene have been proven to be the cause of SWS and port-wine stains. [15] Knowledge of these diseases is still in the process of accumulation and is incomplete, but new disease classifications based on pathophysiology and genetics may be developed with further research. In such case, the uncertainties of current clinical diagnosis is expected to improve with subsequent development of more effective treatment strategies based on specific pathophysiology.
Our reported case is unique in that there was diffuse stenosis of multiple arteries as well as venous-capillaries. Current literature explains the pathophysiology of SWS and nevus flammeus as focal venous malformations and according secondary changes.[8] This patient was unique in that the arteries themselves were extensively involved throughout the body, without being limited to a dermatome or venous drainage passageway. This cannot be sufficiently explained with the current pathophysiological mechanism, and further studies are warranted.
Another key point to consider in this case is the near-fatal rhabdomyolysis which was severe enough to lead to CPR. Rhabdomyolysis may occur secondary to CPR, but laboratory test results sampled immediately after initiation of CPR showed severe hyperphosphatemia (26.9 mg/dL), suggesting rhabdomyolysis as a possible cause of CPR rather than a secondary result. Rhabdomyolysis in SWS is extremely rare with only one case reported to date. In the sole case report, there were myopathological features on muscle biopsy and a high level of long-chain acyl-carnitine on blood testing, and the cause was explained in the perspective of lipid metabolic myopathy.[16] However, there have been no reports of rhabdomyolysis occurring due to hypoperfusion of muscles stemming from stenosis of multiple arteries. In the current case, arterial stenosis was not an acute event but was present chronically, and thus the interaction of additional trigger factors with arterial stenosis may have resulted in rhabdomyolysis. Generally, the known causes of rhabdomyolysis include hypoxic, physical, chemical, and biologic injury.[17] Among these, this patient may have been affected by the interplay of hypoxic (arterial stenosis), chemical (medication and electrolyte imbalance), and/or biologic (infectious vasculitis and myositis) causes. The administration of pethidine may have been a factor as there have been reports of rhabdomyolysis occurring after opioid administration.[18] The patient also demonstrated hypokalemia (2.6 mmol/L) which was identified as a cause of rhabdomyolysis in previous reports.[19] Meanwhile, in a recent report, there was a case of rhabdomyolysis following coronary angiography, and the possible links between rhabdomyolysis and contrast media were suggested.[20] Although the initial CRP level was normal at 0.48 mg/dL, it increased upon follow up. CRP may have risen due to the rhabdomyolysis itself, but considering that there was leukocytosis with an initial WBC count of 17,740/mm3, rhabdomyolysis due to infection cannot be ruled out. ESR rose after discontinuing IV dexamethasone on the 19th day of hospitalization, and deflazacort was started thereafter which suggests that inflammatory processes such as vasculitis may be involved.
Follow-up of CT angiography after two weeks of anti-inflammatory treatment showed improved arterial narrowing of the lower extremities, an indication that the main cause of arterial narrowing may have been an inflammatory process such as vasculitis. The attenuation in arterial stenosis with short-term anti-inflammatory therapy implies that the process was also acute. However, it must be acknowledged that the patient had complained of symptoms such as intermittent left sided weakness for several years. There is a limit in explaining the cause of narrowing as having resulted from an acute inflammation and it seems more valid to consider the inflammatory process as a possible trigger factor for further narrowing of arteries on a preexisting background of chronic diffuse vascular stenosis.
We report a case of renovascular hypertension and severe rhabdomyolysis occurring due to predominantly systemic arterial malformation, and the resultant near-fatal complications. It is easy to focus on the skin manifestations in PPV, and on neurologic manifestations in those cases associated with SWS. Considering the severity of complications in this report, one must recognize that patients may manifest with near-fatal rhabdomyolysis without neurologic alteration such as seizure.
Go to :
 XML Download
XML Download