

 PDF
PDF Citation
Citation Print
Print



Introduction
Hyperlipidemia is an important risk factor for coronary and cerebral artery atherosclerosis. However, its impact may differ according to stroke subtype. In the Atherosclerosis Risk in Communities cohort study [1], clinical ischemic stroke was documented in 305 of 14,175 participants over a mean 10-year follow-up period. Multivariable analysis revealed a weak and inconsistent association between low-density lipoprotein cholesterol (LDL-C) concentration and ischemic stroke, which differs from the close association of LDL-C with coronary heart disease. This difference may be attributed to differences in the relationships of LDL-C with various stroke subtypes, for example, large artery atherosclerosis (LAA) versus lacunar infarction (LI). Thus, although randomized controlled trials have shown that reducing circulating LDL-C levels by using statins is associated with a lower recurrent cardiovascular event risk in patients with coronary heart disease [2], the benefits of statin use in patients with stroke should be interpreted with caution.
Indeed, trials suggest that statin use is beneficial in patients with stroke as well. In the Heart Protection Study (HPS) [3], investigators recruited 3,280 adults (aged 40 to 80 years) with stroke and an additional 17,256 with other occlusive arterial diseases or diabetes. The subjects were randomly allocated 40 mg simvastatin daily or a matching placebo. Overall, there was a significant 25% reduction in the first event rate for stroke (444 [43%] simvastatin vs. 585 [57%] placebo; P<0.0001) and no apparent difference in hemorrhagic stroke (51 [0.5%] vs. 53 [0.5%]; rate ratio, 0.95; 95% confidence interval [CI], 0.65 to 1.40; P=0.8). The Stroke Prevention by Aggressive Reduction in Cholesterol Level (SPARCL) trial [4] evaluated the efficacy of statin therapy in patients with stroke who did not have a history of coronary heart disease. In total, 4,732 patients who had experienced stroke or transient ischemic attack (TIA) were randomized to receive either atorvastatin 80 mg or placebo. Atorvastatin use was associated with a 16% reduction in the cardiovascular event risk. In particular, post hoc analysis revealed that, compared with patients who had an increase or no change in LDL-C, patients with a ≥50% reduction in LDL-C level had a 31% reduction in stroke risk (hazard ratio [HR], 0.69; 95% CI, 0.55 to 0.87; P=0.0016) [5].
In the binational (French and Korean) Treat Stroke to Target (TST) study [6], the investigators enrolled patients who had experienced a stroke or TIA and showed evidence of cerebrovascular or cardiac atherosclerosis. They were randomly assigned to the lower target (LDL-C level <70 mg/dL) or higher target group (LDL-C level, 100±10 mg/dL). The primary outcome was a composite of ischemic stroke, myocardial infarction, new ischemic symptoms requiring urgent coronary or carotid/cerebral revascularization, and vascular death. In total, 2,860 patients were enrolled and followed-up for a median of 3.5 years. The mean LDL-C concentrations achieved were 65 and 96 mg/dL in the lower target and higher target groups, respectively. The primary composite endpoint occurred in 121 (8.5%) and 156 (10.9%) participants, respectively (adjusted HR, 0.77; 95% CI, 0.61 to 0.98; P=0.035). Similar results were obtained when French participants were analyzed separately [7]; the primary endpoint occurred in 9.6% and 12.9% of the participants, respectively (HR, 0.74; 95% CI, 0.57 to 0.94; P=0.019). Myocardial infarction or urgent coronary revascularization occurred in 1.7% and 2.5% of the participants, respectively (HR, 0.66; 95% CI, 0.37 to 1.20; P=0.18), and cerebral infarction or urgent cerebral revascularization occurred in 6.7% and 9.1% of the patients, respectively (HR, 0.73; 95% CI, 0.54 to 0.99; P=0.046).
Based on these results, recent guidelines have recommended the use of a high-dose statin with a low LDL-C target in patients with ischemic stroke [8-10]. However, there are concerns that this strategy may not be universally applied to all patients with ischemic stroke. In the HPS trial [3], among participants with prior stroke, there was no apparent reduction in the incidence of recurrent stroke (rate ratio, 0.98; 95% CI, 0.79 to 1.22) in the simvastatin group, even if there was a significant reduction in the rate of any major vascular event (406 [24.7%] vs. 488 [29.8%], P=0001). Although this result may have been attributed to the relatively small incidence of recurrent strokes in patients with prior stroke, this may suggest that the impact of statins on the recurrence of a stroke may not be as great as in the prevention of coronary disease. In the SPARCL trial, further analysis showed that there was a 33% reduction in participants who had internal carotid artery (ICA) disease, whereas statin administration had a minor effect in participants who did not have ICA disease [11], who were likely to have vascular diseases in the more distal cerebral artery, such as intracranial atherosclerosis (ICAS) or small vessel disease (SVD).
Finally, in the TST trial, although the lower target strategy was superior to the higher target strategy in the French population, the benefit of the “lower target” was not evident when South Korean patients were separately analyzed. Three hundred and seven and 355 patients were assigned to the lower target and higher target groups, respectively. After a median 2.0-year follow-up, the average LDL-C levels achieved were 66 and 96 mg/dL in the lower target and higher target groups, respectively. Preliminary analysis showed neutral results for the primary endpoint. Although the “lower target” strategy was clearly more favorable in the prevention of coronary diseases, it apparently was less effective than the “higher target” strategy in the prevention of stroke and TIA (unpublished data).
Considering that extracranial atherosclerosis (ECAS) is less common in Asian than in French subjects, this difference may be attributed to the lower incidence of ICA disease and a higher incidence of ICAS or SVD among South Koreans than their French counterparts. However, the results should be cautiously interpreted given the relatively small outcomes and shorter follow-up period in the South Korean than in the French counterparts (median 2.0 years vs. 5.3 years, respectively). Therefore, the South Korean extension study is currently ongoing with an estimated average follow-up duration of 5.0 years.
The purpose of this narrative review was to describe the role of blood lipids as a risk factor for various subtypes of non-cardioembolic stroke. The differences in the efficacy of lipid-lowering therapy on various stroke subtypes are also described. Because most lipid-lowering agents studied to date are statins, this review will primarily focus on statin therapies.
Go to : 
Small vessel disease
SVD manifests as white matter hyperintensities (WMHs), LI, cerebral microbleeds (CMBs), or intracerebral hemorrhage (ICH).
Hemorrhagic small vessel disease (intracerebral hemorrhage)
Role of blood lipid in hemorrhagic small vessel disease
Although hyperlipidemia is an important risk factor for coronary heart disease and ischemic stroke, it may protect individuals from developing ICH. A study in the United States of America (USA) examined the relationship between the serum total cholesterol level and risk of death from a stroke during a 6-year follow-up in 350,977 men aged 35 to 57 years. They found that the 6-year risk of death from ICH was three times higher in men with serum total cholesterol levels <160 mg/dL [12]. A South Korean study also showed that hypocholesterolemia was associated with an increased ICH risk. This study additionally revealed that the ICH risk in subjects with hypocholesterolemia was more augmented in those with concomitant hypertension [13]. Another South Korean case-control study showed that decreased fat intake was an important factor for ICH development in men [14]. A recent meta-analysis [15] involving 12 prospective studies with 476,173 participants showed that a 10 mg/dL increase in LDL-C was associated with a 3% lower ICH risk (pooled relative risk [RR], 0.97; 95% CI, 0.95 to 0.98). The association appeared to be more pronounced in Asians (pooled RR, 0.95; 95% CI, 0.92 to 0.98) relative to Caucasians (pooled RR, 0.98; 95% CI, 0.97 to 1.00). Decreased serum lipid levels were also found to be an independent risk factor for CMBs [16]. Thus, hypolipidemia appears to be a risk factor for brain microbleeds or macrobleeds, particularly in patients with hypertension. Differences in serum cholesterol levels among patients with different types of stroke were also demonstrated in an autopsy study from Japan [17], with mean serum cholesterol levels of 164±2, 177±3, and 200±4 mg/dL for patients with ICH, SVD, and cortical infarction, respectively. These data appear to be related to the degree of underlying vascular atherosclerosis among the groups; the mean values of atherosclerosis scores (assigning grade 0 for no stenosis to 4 for ≥50% stenosis in each of the 22 sites of the basal cerebral arteries) were 25±1, 35±2, and 40±2, respectively.
Lipid-lowering therapy in patients with intracerebral hemorrhage
Since the SPARCL study showed that patients on statin treatment had a significant increase (HR, 1.66; 95% CI, 1.08 to 2.55) in the ICH risk [4], there had been concerns regarding ICH development in patients taking statins. In 2011, a meta-analysis involving 23 randomized trials and 19 observational studies was published [18], which showed that statin use was not associated with an increased ICH risk in randomized trials (RR, 1.10; 95% CI, 0.86 to 1.41), cohort studies (RR, 0.94; 95% CI, 0.81 to 1.10), or case-control studies (RR, 0.60; 95% CI, 0.41 to 0.88). Another meta-analysis published a year later included 91,588 and 91,215 subjects in the statin treatment and control groups, respectively [19]. Although total stroke (odds ratio [OR], 0.84; 95% CI, 0.78 to 0.91; P<0.0001) and all-cause mortality (OR, 0.92; 95% CI, 0.87 to 0.96; P=0.0007) were significantly reduced in the statin therapy group, there was no significant difference in the ICH incidence between the groups (OR, 1.08; 95% CI, 0.88 to 1.32; P=0.47). ICH risk was not related to the degree of LDL-C reduction or achieved LDL-C level.
Subsequently, a large population-based Swedish study was published that included 7,696 ICH cases and 14,670 age-sex-matched stroke-free controls [20]. After adjusting for vascular risk factors and the use of antithrombotic therapy, statin use before ICH was associated with a decreased ICH risk in patients (OR, 0.68; 95% CI, 0.63 to 0.74). This protective mechanism was thought to be due to the pleiotropic effects of statins, such as anti-inflammatory and antithrombotic properties that may protect the cerebral vessels [21]. A more recent meta-analysis showed that patients who used statins before ICH had a decreased mortality risk at 3 months after symptom onset (OR, 0.47; 95% CI, 0.32 to 0.68) and an increased probability of good functional outcomes (OR, 1.49; 95% CI, 1.01 to 2.19), as compared with those who did not [22]. Therefore, although hypocholesterolemia appears to be a risk factor for ICH, particularly in those with hypertension, statin use does not appear to increase the ICH risk in the normal population and may ameliorate the severity of ICH if it develops.
A recent meta-analysis examined the relationship between statin use and the development of CMBs [23]. It included seven studies (n=3,734 participants, with an unselected general population [n=1,965], patients with ischemic stroke [n=849], patients with hemorrhagic stroke [n=252], and patients with hypertension aged >60 years [n=668]). The results showed that statin use was not associated with CMBs in either unadjusted (OR, 1.15; 95% CI, 0.76 to 1.74) or adjusted analyses (OR, 1.09; 95% CI, 0.64 to 1.86). Statin use was more strongly related to lobar CMB presence (OR, 2.01; 95% CI, 1.48 to 2.72) in the unadjusted, but not in the adjusted analysis (OR, 2.26; 95% CI, 0.86 to 5.91). Thus, statin treatment did not appear to cause CMBs overall.
Unfortunately, there have been no randomized controlled trials examining the efficacy or hazards of lipid-lowering therapy in patients with ICH. A retrospective study from Taiwan, which used the National Health Insurance Research Database data, compared the outcomes of early statin users (n=749) and statin non-users (n=7,583). During follow-up, the incidence of recurrent ICH was similar between the two groups: 9.2% and 8.9% in the statin and control groups, respectively. However, the mortality rate was significantly lower in patients in the statin group: 90 (12.0%) and 1,519 (20.0%) in the statin and control groups (adjusted HR, 0.742; 95% CI, 0.598 to 0.919), respectively [24]. Another study from China that used data from the China National Stroke Registry analyzed 3,218 admitted patients with ICH; 220 (6.8%) were on statins during their hospitalization. Multivariate analysis showed that patients with ICH on statins had better functional outcomes at 3 months (OR, 2.24; 95% CI, 1.49 to 3.36) and at 1 year (OR, 2.04; 95% CI, 1.37 to 3.06), and lower mortality rates at 3 months (OR, 0.44; 95% CI, 0.22 to 0.87) and 1 year (OR, 0.49; 95% CI, 0.27 to 0.86) than those without statin therapy [25].
A retrospective study from Japan [26] involved 381 patients with ICH, among whom statins were used in 56 patients. Serum total cholesterol and LDL-C levels were significantly lower in the statin group than in the non-statin group. Hematoma volume was inversely correlated with total cholesterol and LDL-C levels in both groups. Multiple logistic regression analysis showed that serum cholesterol levels ≤150 mg/dL were associated with worsening of the National Institutes of Health Stroke Scale (NIHSS) score (OR, 1.4; 95% CI, 1.21 to 1.63) and increased hematoma volume (OR, 1.1; 95% CI, 1.07 to 1.13) in patients with ICH. Similar findings were observed when patients who used statins were analyzed separately. A more recent retrospective Finnish study [27] analyzed 964 patients with ICH and reported that, after adjusting for factors such as age, NIHSS, Glasgow Coma Scale, ICH volume, and intraventricular hemorrhage, lower LDL-C levels were independently associated with in-hospital mortality (OR, 0.54; 95% CI, 0.31 to 0.93; P=0.028).
Thus, statin use after ICH may not increase the risk of further ICH and may improve the functional outcome and mortality in patients with ICH. However, excessive lowering of cholesterol levels by statins may have to be avoided. However, these retrospective studies have limitations. Details were frequently unknown, such as why and when the statin was initiated, the type and dose of statin, and the duration of treatment. Prospective, controlled trials are needed to elucidate whether statin therapy is needed in patients with ICH, and if so, when and with what dosage should be initiated.
Ischemic small vessel disease
White matter hyperintensities
(1) Role of blood lipid and white matter hyperintensities
The results of studies on WMH should be interpreted cautiously for the following reasons: First, WMH may not always be an ischemic lesion but can be due to demyelinating diseases or degenerative gliosis. Second, hyperlipidemia may play a role in the development of LAA, and resultant brain hypoperfusion may lead to WMH. Thus, its effect on the underlying large cerebral artery may, in turn, affect the presence of WMH. Third, WMH development may be associated with certain genetic factors, the prevalence of which may differ across ethnicities [28]. These limitations should be kept in mind when interpreting the results of WMH studies.
While WMHs are known to be associated with old age and hypertension [29], the relationship between blood lipids and WMH remains unclear [30]. In a case-control study from China involving 333 and 503 patients with and without WMH, respectively [31], risk factors such as age, smoking, diabetes, and hypertension, but not hyperlipidemia, were associated with the prevalence of WMH. In another study that analyzed two independent hospital-based ischemic stroke cohorts (Massachusetts General Hospital in Boston, USA and Hospital del Mar in Barcelona, Spain), the only independent risk factor clearly related to WMH was hyperlipidemia; patients with hyperlipidemia had significantly (P<0.01) less severe WMH in both cohorts [32]. The Austrian Stroke Prevention Study also showed that age and hypertension were positively associated with the WMH degree, whereas the serum cholesterol level was negatively associated [33]. Thus, similar to ICH, hypocholesterolemia appears to be associated with WMH, while hyperlipidemia may be protective.
(2) Lipid-lowering therapy in patients with white matter hyperintensities
In the Cardiovascular Health Study (CHS), a population-based longitudinal study, 1,919 participants underwent two magnetic resonance imagings (MRIs) at approximately 5-year intervals. Overall, 538 (28%) patients showed worsening of WMH by one grade in most patients (85%). The study analyzed the risk factors associated with WMH worsening separately in patients with initially low-grade (grade 0-1) WMH and those with a higher initial grade. Age, high diastolic blood pressure, and decreased LDL-C levels were associated with worsening of WMH. Although statin use was not related to WMH changes in general, it was associated with an increased WMH progression risk in patients with initially high-grade WMH [34].
In the Prospective Study of Pravastatin in the Elderly at Risk study [35], investigators performed repeated brain MRIs to examine the effect of pravastatin 40 mg/day on the progression of WMH volume. After a mean treatment period of 33 months, WMH volume increased by 1.1 cm3 in both the 270 placebo-treated and 265 pravastatin-treated subjects, reflecting no difference between the two treatment groups. The Cardiovascular Risk Factors and Aging and Incidence of Dementia MRI substudy [36] showed that hyperlipidemia at mid-life was not associated with late-life WMH. However, statin use at mid-life appears to be associated with decreased late-life WMH. Unfortunately, the study was underpowered because of the small number of enrolled patients (n=112).
The Regression of Cerebral Artery Stenosis study was a randomized, double-blind, placebo-controlled study that primarily evaluated the effects of statins on middle cerebral artery (MCA) stenosis progression among stroke-free individuals. Two hundred and eight subjects were randomly assigned to either the placebo (n=102) or simvastatin 20 mg daily (n=106) group. After 2 years of follow-up, there was no significant change in WMH volume between the statin and placebo groups as a whole. However, in individuals with severe baseline WMH, the median volume increase was less in the statin group (1.9 cm3) than in the placebo group (3.0 cm3; P=0.047). Their results were at odds with the CHS study results described above.
Thus, it appears that mid-life hypocholesterolemia is associated with late-life WMH. However, the relationship between statin use and late-life WMH remains unclear. As statin use is closely related to hyperlipidemia, the association between serum cholesterol and WMH may be confounded by statin use. Additionally, statins may have two faces: they may increase late-life WMH by decreasing serum cholesterol but may prevent WMH via other pleiotropic effects. Finally, as addressed above, statins may improve cerebral perfusion by preventing underlying LAA, which may, in turn, prevent late-life WMH.
Lacunar infarcts
(1) Heterogeneity of so-called lacunar infarction
Before we discuss the role of blood lipid or lipid-lowering therapy in patients with an LI, we need to consider the heterogeneous nature of LIs that has not been appropriately considered in previous studies on lipids.
LI, a small, deep infarction, is caused by deep perforating arterial disease and is pathologically characterized by lipohyalinosis or fibrinoid degeneration rather than by significant lipid deposits in the vessel wall. However, recent studies have shown that the pathology of “LI,” defined using imaging criteria, may be heterogeneous. Local thrombi or atheromas in patients with ICAS may obliterate the orifice of the perforator and produce LI associated with clinical “lacunar syndromes”; this is called branch atheromatous disease (BAD) [37]. Imaging methods such as magnetic resonance angiography (MRA) and computed tomographic angiography (CTA) can identify such cases. High-resolution vessel wall MRI (HR-MRI) may be needed to identify mild and diffuse atherosclerosis in intracranial arteries that may not be detected by conventional MRA or CTA [38]. Because the so-called BAD or “branch occlusion” is essentially an LAA, this condition is likely to be associated with atherosclerosis rather than with classical SVD pathologies.
Consequently, Nah et al. [39] investigated whether indicators of SVD and atherosclerosis in patients with LI (subcortical or brainstem infarct of diameter ≤20 mm) differed according to the lesion location and presence of parent artery atherosclerotic disease (PAD). They assessed 449 patients with LI within 48 hours of stroke onset. The LIs were characterized as proximal LIs (pLIs) if the lesions abutted the main artery and distal LIs (dLIs) if they did not. The LIs were also classified according to the lesion location and presence of PAD: (1) pLI with PAD (pLI+PAD); (2) pLI without PAD (pLI–PAD); and (3) dLI without PAD (dLI–PAD) (Figure 1). The prevalence of SVD indicators (WMH and CMB) and atherosclerosis (cerebral atherosclerosis and coronary heart disease) were compared among the groups. The pLI+PAD group had the highest prevalence of atherosclerosis indicators and lowest prevalence of SVD indicators, whereas the dLI–PAD group had the lowest prevalence of atherosclerosis indicators and highest prevalence of SVD indicators. The pLI–PAD group showed intermediate features. These results suggest that LI pathogenesis is heterogeneous and varies according to the lesion location and presence of PAD. Additionally, compared with pLI, dLI was marginally associated with hypertension (P=0.08) but was less frequently associated with diabetes (P=0.003). In their study, the prevalence of hyperlipidemia did not differ among the groups. However, in a similar study conducted in another part of Asia [40], LDL-C was more closely associated with pLI than with dLI in older patients (aged >65 years). In another South Korean study, the authors found that, when stroke recurred, patients with pLI were more likely to develop atherosclerotic cerebral infarction than those with dLI [41], again suggesting that pLI may have more atherosclerotic characteristics than dLI.
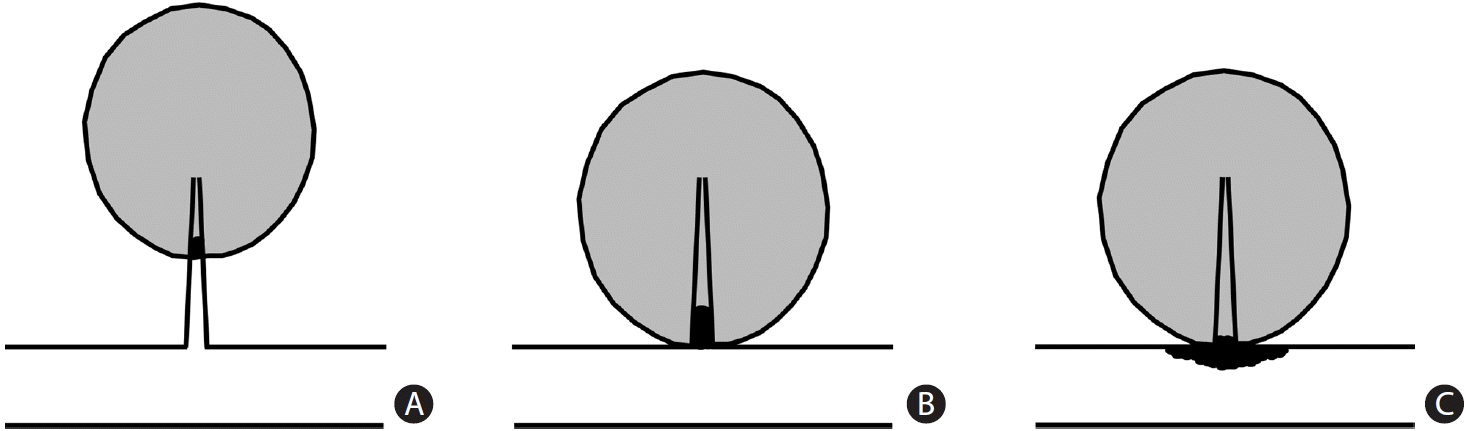 | Figure 1.Classification of lacunar infarction (LI) according to the location of the lesion and presence/absence of parental arterial disease (PAD). (A) Distal LI without PAD, (B) proximal LI without PAD, (C) proximal LI associated with PAD. Adapted from Nah et al.[39]
|
(2) Role of blood lipid and lacunar infarctions
In a cross-sectional study conducted in China [42], the investigators analyzed the clinical characteristics of 1,982 patients with ischemic stroke. The stroke subtypes were classified as LAA, LI, cardioembolism, or undetermined, and 1,207, 566, 173, and 36 of the participants were diagnosed with each of these subtypes, respectively. Multivariate analysis showed that hypertension (OR, 1.832) and WMH (OR, 1.865) were more strongly associated with LI than LAA, whereas LDL-C levels (OR, 0.774) were more strongly associated with LAA than with LI. In another study [43], the investigators compared the risk factors in 573 participants with a single LI with those of 122 participants with recurrent LIs. They found that hypertension was more prevalent (81% vs. 70%, respectively; P=0.01), whereas hyperlipidemia was less prevalent (15% vs. 24%; P=0.025) in the recurrent-LI group than in the single-LI group. The authors suggested that hypertension may be a risk factor for LI recurrence, whereas hyperlipidemia might reduce this risk.
(3) Lipid-lowering therapy in patients with lacunar infarction
Several Asian studies have investigated the possibility that the effect of lipid-lowering therapy may differ according to the stroke subtype. In the randomized Japan Statin Treatment Against Recurrent Stroke (JSTAR) [44] study, patients aged 45 to 80 years with a history of non-cardiogenic ischemic stroke within the preceding 1 month to 3 years were enrolled at 123 centers. A total of 1,578 patients were randomly allocated to the pravastatin (10 mg/day) or control group. Pravastatin treatment reduced stroke recurrence in patients with LAA (HR, 0.33; 95% CI, 0.15 to 0.74; P=0.0047), but not in those with other stroke subtypes. In patients with LI, there was a tendency for increased stroke occurrence in the statin group, although this was not statistically significant.
On the other hand, a retrospective study from Spain [45] assessed 2,742 patients with stroke, among whom 281 patients (10.2%) were pre-stroke statin users. Logistic regression analyses showed that statin treatment was an independent predictor for better functional outcome at discharge among all patients with stroke (OR, 2.08; 95% CI, 1.39 to 3.1), those with LAA (OR, 2.79; 95% CI, 1.33 to 5.84), and those with LI (OR, 2.28; 95% CI, 1.15 to 4.52). The authors hypothesized that statins might improve cerebral endothelial function in these patients.
Thus, hyperlipidemia does not play an important role in the development of LI compared with LAA, and the effect of lipid-lowering therapy on LI remains unclear. As discussed above, it should be remembered that LI is a heterogeneous condition; among patients with LI, pLI may be more closely associated with hyperlipidemia and may respond better to statin therapy than dLI. Further studies are needed to determine whether lipid-lowering therapy is effective, at least in patients with a certain subtype of LI.
Intracranial atherosclerosis
Role of blood lipid in intracranial atherosclerosis
Similar to atherosclerosis in other vessels, ICAS is caused by vascular risk factors, such as age, hypertension, diabetes mellitus, cigarette smoking, and hyperlipidemia. However, studies that compared the risk factors between ICAS and ECAS have shown that hyperlipidemia is a less important risk factor for ICAS than for ECAS [46-51]. To the best of my knowledge, no studies have reported the opposite results. Because Asians have lower serum cholesterol levels than Caucasians, this difference may at least in part explain the ethnic differences in the location of cerebral atherosclerosis. Intracranial arteries may have an inherent resistance to the toxic effects of hypercholesterolemia. In monkeys [52] and rabbits [53], the development of atherosclerosis after consuming an atherogenic diet occurred significantly later in intracranial arteries than in extracranial arteries. It has been suggested that a specific glycocalyx composition on luminal endothelial cells might inhibit trapping of chylomicrons and very-low-density lipoprotein, resulting in reduced deposition of apolipoproteins in the intima of intracranial vessels [52].
Although there may be differences in the pathological features between ICAS and ECAS, few studies have explored this issue. A recent autopsy study from Hong Kong [54] examined the pathology of large intracranial arteries in 32 autopsy cases aged ≥45 years. Although atherosclerotic lesions were present in 91 (71%) of the 128 arteries, features of complicated plaques were infrequent; plaque hemorrhage was encountered in 12%, neo-vasculature in 12%, lumen thrombi in 13%, macrophage infiltration in 20%, and calcification in 25% of arteries. Luminal narrowing was the most severe in the MCA, followed by the vertebral artery (VA), while the basilar artery (BA) was the least stenotic. Thus, it appears that the lesions generally lacked features of complicated plaques in proximal ICA atherosclerotic disease. However, it should be kept in mind that the complicated plaques described in proximal ICA disease were obtained primarily in patients who underwent carotid endarterectomy, a highly selected patient population mostly with severe atherosclerosis, whereas most of the subjects in the autopsy study described above did not have a history of stroke.
Lipid-lowering therapy in patients with intracranial atherosclerosis
There have been few studies on lipid-lowering therapies that exclusively focus on ICAS. In a recent study [55], the investigators enrolled statin-naïve patients with ischemic stroke who had symptomatic ICAS (>50% stenosis) in the proximal portion of the MCA, BA, or intracranial portion of the ICA. Atorvastatin (40 to 80 mg) or rosuvastatin (20 mg) was administered for 6 months, and patients underwent HR-MRI before and after statin treatment. Pre-specified endpoints that were measured after 6 months of statin treatment were: (1) degree of stenosis, (2) remodeling index, (3) wall area index, and (4) enhancement volume of atherosclerotic plaque on HR-MRI. The LDL-C concentrations of the 77 enrolled patients at the initial and follow-up assessments were 125.8±35.7 and 61.0±19.3 mg/dL, respectively. Overall, statin treatment significantly reduced plaque accumulation (32.07±39.15 mm3 vs. 17.06±34.53 mm3, P=0.013), wall area index (7.5±4.3 vs. 5.9±4.1, P=0.016), and degree of stenosis (76.5%±20.2% vs. 64.1%±21.3%, P<0.001), but not the remodeling index (P=0.195). This suggests that high-dose statins may benefit patients with symptomatic ICAS. However, this study has important limitations. First, it was uncertain whether such MRI changes could translate into clinical benefits. Second, because there was no control group, it remains unclear whether the MRI changes were indeed due to statin administration. Third, it is unknown whether the dose used in this study was the most appropriate for patients with ICAS. It is noteworthy that despite intensive statin therapy, 35% of the participants showed no beneficial changes; they showed unchanged or worsened vessel pathologies.
To the best of my knowledge, there is only one controlled study on statins for asymptomatic ICAS. In a single-center prospective study [56], 71 asymptomatic patients with ICAS were treated with rosuvastatin, and their vascular stenoses were evaluated using transcranial color-coded sonography (TCCS) before and after treatment. The therapeutic target was a reduction in LDL-C concentration of ≤1.8 mmol/L or a ≥50% reduction from baseline over 2 years. The participants were allocated to an intensive statin treatment (IST) or standard statin treatment (SST) group. One hundred and four ICASs were identified in 51 participants and 47 in 20 in the IST and SST groups, respectively. After 1 year, there was a larger decrease in LDL-C concentration in the IST than in the SST group (1.5±0.3 mmol/L vs. 2.2±0.6 mmol/L, respectively; P≤0.001). The percentage regression of the ICAS lesions in the IST group tended to be greater than that in the SST group (26.3% vs. 5.9%, P=0.052). Forty-nine lesions in 25 participants and 16 lesions in seven in the IST and SST groups, respectively, were reassessed 2 years later. At this time, the LDL-C concentration had decreased more in the IST group than in the SST group (1.6±0.3 mmol/L vs. 2.46±0.8 mmol/L, P=0.048), and the percentage regression of ICAS lesions in the IST group was significantly greater than that in the SST group (34.7% vs. 6.3%, P=0.017). This result suggests that the degree of stenosis in ICAS can be ameliorated using intensive statin therapy. However, this study had limitations: the number of participants who were followed for 2 years was too small, and the stenosis was assessed by TCCS rather than by standard methods, such as MRA, CTA, or conventional angiography.
Thus, although statins probably benefit patients with ICAS, further studies including a control group, assessment of clinical endpoints, and comparison of the effects of statin therapy in patients with ICAS and ECAS are required. Changes in vessel stenosis or plaque volume should also be assessed more meticulously. Considering the heterogeneity of ICAS, other issues should be considered in future trials. First, strokes in patients with ICAS are caused by various mechanisms, including atherosclerotic steno-occlusion, BAD leading to perforator occlusion, artery-to-artery embolism, and hemodynamic impairment [37,57]. Recent studies have shown that risk factors and vessel wall pathology may differ among different mechanisms [39,58-62]. Specifically, rupture-prone positive remodeling and enhancing plaques are more frequently observed in non-BAD-type ICAS than in BAD-type ICAS [62]. Therefore, it may be assumed that lipid-lowering agents might be more effective in patients with non-BAD type ICAS. Second, as imaging techniques, such as MRA and catheter angiography, can examine luminal stenosis but not vascular wall pathology, non-atherosclerotic etiologies, such as Moyamoya disease, dissection, and vasculitis can be miscategorized as ICAS. One diagnostic tool that can be used to examine the vessel wall is HR-MRI [63]. A study from South Korea [64] assessed HR-MRI findings in 95 young (≤55 years) patients who were diagnosed with presumable ICAS. They found that only 26 (27.4%) had HR-MRI findings consistent with atherosclerosis, whereas others showed findings consistent with other vascular pathologies. These data suggest that diagnosis based solely on angiogram findings may not be accurate, which may explain why the effect of statins was not consistent in patients with ICAS [55].
Extracranial atherosclerosis
As discussed above, hyperlipidemia is an important risk factor for ECAS, probably more so than in ICAS. Thus, the effects of lipid-lowering agents should also be robust under these conditions.
Proximal internal carotid artery disease
Proximal ICA atherosclerosis has many similarities to coronary artery disease, and hypercholesterolemia is an important risk factor. Of the 4,731 SPARCL participants, a subset of 1,007 participants had ICA disease. In this subgroup, atorvastatin therapy was associated with a 33% reduction in the risk of recurrent stroke, which was far greater than the 16% reduction observed in all SPARCL participants [11]. Thus, among patients with stroke, those with ICA disease appear to benefit most from a high dose of statin.
Statins appear to influence the composition of the carotid plaques. A recent meta-analysis of seven studies (361 patients) [65] included prospective studies using serial HR-MRI in patients on statin therapy to document changes in carotid plaque composition. Outcomes included changes in lipid-rich-necrotic-core (LRNC) volume, wall volume, and lumen volume at varying time points after initiation of statin therapy. There was no significant change in carotid wall volume or lumen volume at any time point. While there was no significant difference in LRNC before 7 to 12 months after statin initiation, there was a significant decrease in LRNC volume (weighted mean difference –9.9 mm3; 95% CI, –8.9 to –2.3) at >12 months.
Subsequently, a population-based Rotterdam Study [66] enrolled 1,740 participants with ICA disease who underwent carotid artery MRI to determine the presence of calcification, lipid core, and intra-plaque hemorrhage. Statin treatment was associated with a higher presence of calcification (OR, 1.73; 95% CI, 1.22 to 2.44), which was more apparent in patients with longer statin use. Current statin treatment was also associated with a lower presence of lipid core (OR, 0.66; 95% CI, 0.42 to 1.04), but only when statins were used for ≤10 months. Although there are subtle differences in the results, the two studies suggest that statin therapy favorably influences the composition of carotid atherosclerosis by shifting the composition from vulnerable plaque with a lipid core to a more stable calcified plaque.
A more recent Treat Stroke to Target–Plaque Ultrasound Study study [67] assessed the serial ultrasonic carotid findings for a median follow-up of 3.1 years. Patients were randomized to the lower LDL-C target (mean, 64 mg/dL; n=201) or higher LDL-C target group (mean, 106 mg/dL; n=212). Compared with the higher-target group, the lower-target group had a similar incidence of newly diagnosed carotid plaque: 46/201 (5-year rate, 26.1%) versus 45/212 (5-year rate, 29.7%). The change in common carotid arteries intima-media thickness was significantly (P=0.004) better in the lower-target group (−10.53 μm; 95% CI, –14.21 to –6.85) than in the higher target group (−2.69 μm; 95% CI, –6.55 to 1.18). Thus, in this study, statin use with an LDL-C target of <70 mg/dL did not reduce the incidence of new carotid plaques but produced greater regression of carotid atherosclerosis than an LDL-C target of 90 to 110 mg/dL.
The benefits of statins in patients who underwent carotid revascularization have also been investigated. A population-based, retrospective cohort study in Ontario, Canada, evaluated the statin effect in individuals aged ≥66 years who underwent carotid endarterectomy or stenting during 2002-2014 [68]. The study primarily assessed the effect of pre-procedural statin for the composite of stroke, myocardial infarction, or death at 1 year. Additionally, the benefit of persistent long-term statin use was assessed 5 years after the procedure. Of the 10,723 patients identified, 7,893 (73.6%) were pre-procedural statin users. Pre-procedural statin use was associated with a 24% reduction (adjusted HR, 0.76; 95% CI, 0.70 to 0.83) in the risk of the 1-year composite of stroke, myocardial infarction, or death. At 5 years, persistent long-term statin use was associated with a 25% risk reduction (adjusted HR, 0.75; 95% CI, 0.71 to 0.80) for the composite outcome. These results suggest that statins should be used in patients undergoing carotid revascularization.
Vertebral artery disease
Unlike ICA, VA atherosclerosis has rarely been examined in clinical studies of lipid-lowering agents. Similar to ICA, VA atherosclerotic disease produces artery-to-artery embolism, leading to posterior circulation ischemia. Therefore, the plaque-stabilizing effect of statins may be equally important in patients with VA disease. Thus, statins have been recommended in patients with atherosclerotic VA disease [69]. However, although pathological studies are scarce owing to the fact that endarterectomy is rarely performed in VA disease, plaque ulceration and hemorrhage are considered to be less common in VA atherosclerosis than in ICA disease; the lesions are more often fibrous and smooth [70]. Thus, it remains uncertain whether statins are really effective, and if so, what the optimal LDL-C target would be for patients with VA atherosclerotic disease.
Go to :
Comments on recent guidelines
The 2018 American College of Cardiology (ACC)/American Heart Association (AHA) task forces reported guidelines on the management of blood cholesterol [71], which were: (1) in patients with clinical atherosclerotic cardiovascular disease (ASCVD), reduce LDL-C with high-intensity statin therapy or maximally tolerated statin therapy. The more LDL-C is reduced, the greater the subsequent risk reduction. (2) In very high-risk ASCVD, use an LDL-C threshold of 70 mg/dL and consider the addition of non-statins to statin therapy. Very high-risk ASCVD includes a history of multiple major ASCVD events, one major ASCVD event, and multiple high-risk conditions. In this paper, the authors appropriately used the term “atherosclerotic” disease. Thus, if ASCVD includes stroke, it should mean an “atherosclerotic” stroke. In that regard, this is a reasonable guideline, although the author still doubts whether the same principle can be applied to ICAS.
However, in subsequent guidelines, the term “atherosclerotic” was often omitted. One example was a guideline for the management of patients with “acute ischemic stroke (AIS)” published in 2019 [8]. Authors wrote that patients with AIS should be managed according to the 2018 ACC/AHA lipid management guidelines, as described above. The second example is a guideline by the Hellenic Stroke Organization and Hellenic Atherosclerosis Society [9]. Authors reported that (1) patients with ischemic stroke or TIA should receive lipid-modifying treatment with a high-intensity statin (grade of recommendation/level of evidence, 1A) and (2) patients with ischemic stroke or TIA should be treated with an LDL-C target <55 mg/dL and at least 50% reduction of baseline LDL-C levels (1B). Finally, the European Stroke Organization (ESO)-Karolinska guideline [10] reported that “Statins should be used as a part of secondary prophylactic treatment after an ischemic stroke or TIA.” Most benefits were observed with atorvastatin 80 mg (Grade A). Aggressive intensive lipid-lowering therapy with statins (sometimes with additional ezetimibe) reduces the risk of stroke in stroke survivors in an LDL-C-dependent manner (Grade A).
These recommendations were based on the TST, JSTAR, and SPARCL trials [3,4,6,44] that we have already discussed. In these guidelines, “ischemic stroke” should be replaced with “atherosclerotic ischemic stroke” for the following reasons. First, TST did not include general stroke patients, but only those with evidence of cerebral/cardiac vessel atherosclerosis. Second, in the JSTAR trial, pravastatin therapy reduced stroke risk in patients with LAA but not in those with LI. Third, in the SPARCL trial, the benefit of statins was minimal in patients without ICA disease, and (4) the term “ischemic stroke” broadly includes non-atherosclerotic diseases such as cardioembolic stroke, dissection, vasculitis, Moyamoya disease, and stroke of unknown etiology. Although the ESO-Karolinska guideline [10] cautiously wrote that statin use in the secondary prevention of ischemic stroke caused by less frequent non-atherosclerotic etiologies such as arterial dissection and patent foramen ovale requires further investigation, this is not enough. Although a registry study [72] and a hospital study [73] suggested that statin therapy may reduce recurrent stroke or vascular death risk in patients with atrial fibrillation, another study [74] failed to show such a benefit. More importantly, these non-atherosclerotic strokes were excluded from the above trials, and there are no randomized trials that properly examined the benefit of lipid-lowering therapy in patients with stroke associated with atrial fibrillation or other determined or unknown etiologies. Thus, in the guidelines, “high dose statin” and “low LDL-C target” strategies should be limited to patients with atherosclerotic stroke, especially those with ICA disease, such that physicians are not misled.
Go to :
Conclusions
The roles of blood lipids and lipid-lowering therapies differ among stroke subtypes (Table 1). For SVDs, hypolipidemia appears to increase the risk of hemorrhagic stroke, microbleeds, WMH, and probably LI, especially in subjects with hypertension. However, statins do not appear to increase the risk of further stroke in patients with SVD, probably because statins have various pleiotropic effects in addition to the lipid-lowering effect. Although the evidence is not robust, statins may improve functional outcomes or decrease mortality in patients with ICH. Because of the paucity of clinical trials and heterogeneity of pathogenesis, study results on LI are inconclusive, although it appears clear that lipid-lowering therapy is less effective in this condition than in LAA. Although hyperlipidemia is a risk factor for ICAS, its impact is less marked than that of ECAS. Statin use is probably useful in this condition, but the effect may be less robust or inconsistent than in ECAS. Hyperlipidemia is a definitive risk factor for ECAS, and “high dose statin” and “low LDL-C targets” should be a reasonable strategy in this condition. Further studies are needed to elucidate whether lipid-lowering therapy is needed, and if so, what would be the appropriate LDL-C target levels in patients with stroke due to different levels of cerebral artery diseases.
Table 1.
Role of blood lipid and lipid lowering therapy in various subtypes of stroke
| Variable | Role of hyperlipidemia as a risk factor | Preventive effect of lipid lowering therapy | Therapeutic effect* of lipid lowering therapy | |
|---|---|---|---|---|
| Small vessel disease | ||||
| Intracerebral hemorrhage | – | – | + | |
| Microbleeds | – | ? | ? | |
| White matter hyperintensities | – | ± | ? | |
| Lacunar infarction | ± | ? | ± | |
| Large artery atherosclerosis | ||||
| Intracranial atherosclerosis | + | + | + | |
| Extracranial atherosclerosis | ++ | ++ | ++ | |
![]()
Go to :
 XML Download
XML Download