

 ePub
ePub Citation
Citation Print
Print



Autoimmune reactions in connective tissue diseases cause systemic and have a significant impact on the survival of patients with pulmonary manifestations. Dermatomyositis is an incurable connective tissue disease that results from inflammatory and degenerative changes in muscle and skin, it has several prominent symptoms, including skin lesions (e.g. heliotrope erythema, Gottron’s sign, Gottron’s papules) accompanied by erythema and edema, proximal muscle weakness of limbs and nonsuppurative inflammation.[1] The disease can involve the whole body including the respiratory system. Cryptogenic organizing pneumonia (COP) is a common pattern of pulmonary complication known to respond well to steroid treatment. However, when the clinical course in more aggressive and immunosuppressive agents including steroid, azathioprine, mycophenolate, cyclophosphamide and methotrexate are not very effective, immunosuppressive agents such as rituximab and tacrolimus can be used.[2-4] Moreover, immunoglobulin injection has shown efficacy in patients with severe interstitial lung disease (ILD), even though its mechanisms of action in dermatomyositis and polymyositis is not clear.[5] Severe COP with respiratory failure caused by dermatomyositis is very rare, and treatment remains difficult; therefore, it is important to share experience of successful treatment of patients in clinical practice. Using immunosuppressive treatments such as high-dose steroid, cyclophosphamide and intravenous (i.v.) immunoglobulin, we have successfully treated patients admitted to hospital with COP and respiratory failure caused by dermatomyositis. Here, we report such a case and review the literature.
Case Report
A 46-year-old man was admitted to the emergency room with dyspnea that had worsened during the preceding 10 days. He was a current smoker of 5 pack years with no underlying disease. Although he usually had a nonspecific whole-body skin rash and wax-and-wane myalgia, this had not been assessed. At the time of admission, he had rash on both sides of the pelvis and groin. He lives in Los Angeles and a month before had travelled to Las Vegas for about a week. A week after the trip, he began to suffer from fever and chills and was diagnosed with pneumonia in another hospital. He took oral levofloxacin but the symptoms did not improve; furthermore, he continued to suffer from dyspnea. Serologic tests for antinuclear antibodies were positive.
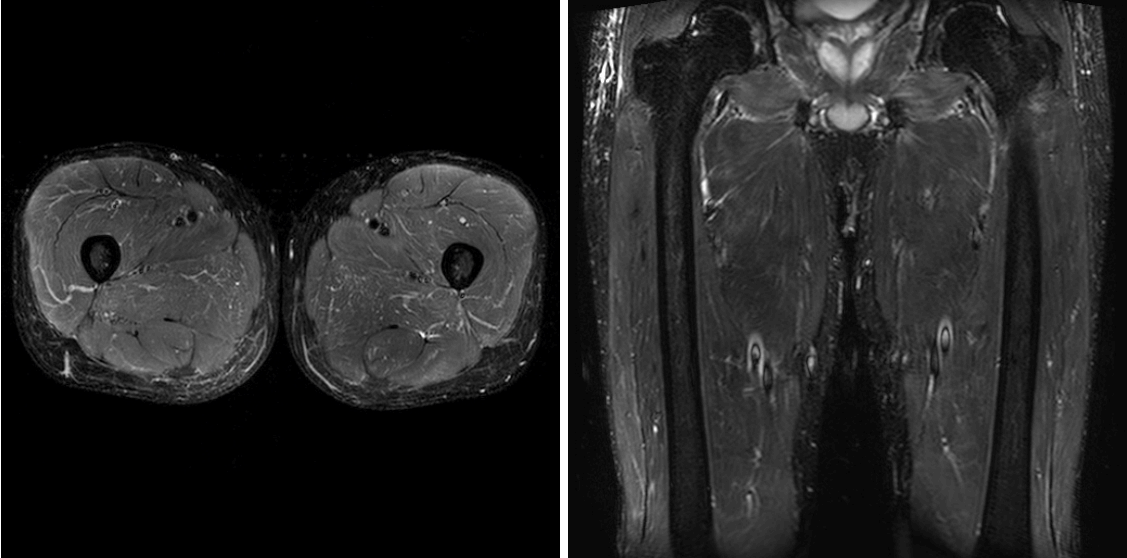
The patient was transferred to our hospital in Seoul with suspicion of rheumatic disease. On admission, his vital signs were as follows: blood pressure 110/50 mmHg, pulse 103 beats/min, respiratory rate 18 breaths/min, body temperature 38.6ºC, with clear consciousness. On physical examination, crackle was heard from the lower lobes of both lungs. The muscular strength of both upper and lower limbs was confirmed as grade 5. There was no sign of tenderness. The patient underwent arterial blood examination while breathing 35% oxygen with a venturi mask and was confirmed to have a PaO2/FiO2 188 mmHg, PaO2 65.8 mmHg, and oxygen saturation 94.1%. The complete blood count showed leukocytes 15,500/mm3 (neutrophils 79.3%), hemoglobin 13.2 g/dL, platelets 229,000/mm3, and erythrocyte sedimentation rate 5 mm/h. Blood chemistry was as follows: blood urea nitrogen 12.6 mg/dL, creatinine 0.92 mg/dL, aspartate transaminase 63 U/L, alanine transaminase 33 U/L, total bilirubin 0.75 mg/dL, lactate dehydrogenase 398 U/L, creatine phosphokinase 1,267 U/L, aldolase 38.0 IU/mL, myoglobin 642 ng/mL, and C-reactive protein 4.97 mg/dL. Rheumatoid factor was 29 IU/mL and antinuclear antibody >1:2560 (cytoplasmic level). A thyroid function test was normal. Anti-dsDNA, anti-Sm, anti-Jo-1, anti-La, anti-RNP, and anti-cardiolipin were all negative; with C3/C4 of 90/35 mg/dL and CH50 of 59.3 U/mL. The anti-Ro antibody test was positive. Chest X-ray showed evidence of consolidation in both lower lobes; and pulmonary infiltration was observed at the margins of both middle lung fields (Fig. 1). Chest computed tomography (CT) gave evidence of organizing pneumonia with peripheral consolidation accompanied by bilateral ground-glass opacity (Fig. 2). Magnetic resonance imaging of the thigh suggested atypical myositis (Fig. 3). In an electromyography test, vastus lateralis and rectus femoris had decreased insertional activity, and motor unit potentials had lower amplitudes and early recruitment patterns. The H-reflex was normal, compatible with myopathy.
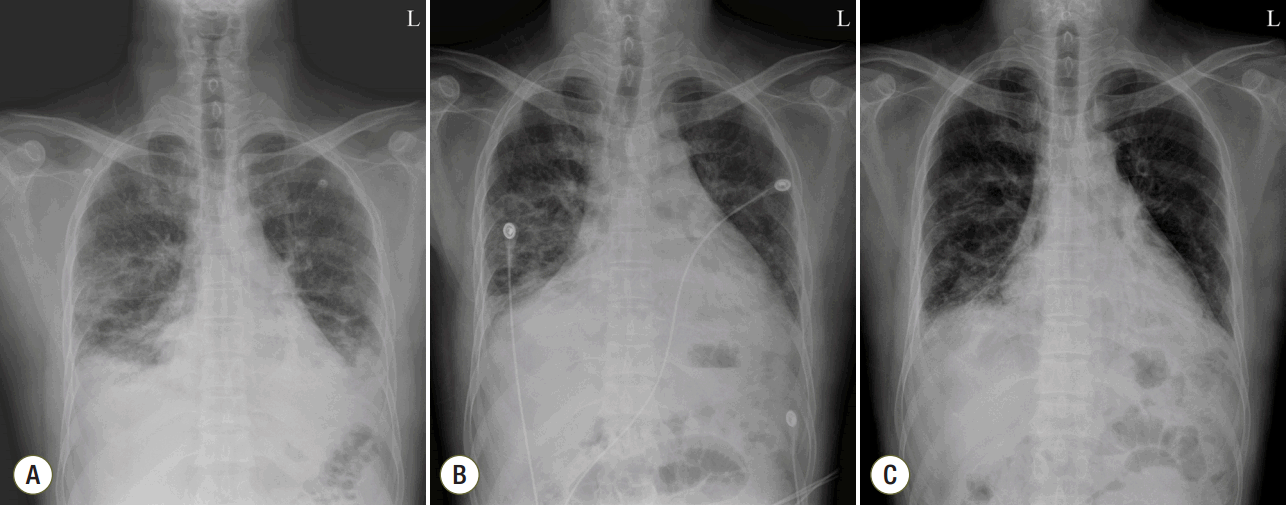 | Fig. 1.Chest X-ray. Chest X-ray on the day of admission, showing an ill-defined consolidation in both lower lobes and infiltration in the peripheral lung in both middle lung fields (A). Chest X-ray on day 22 of hospitalization, showing no improvement of the infiltration in either lower lobe (B). Chest X-ray on day 25 of hospitalization, after administration of intravenous immunoglobulin, showing improvement of the infiltration in both lower lobes (C).
|
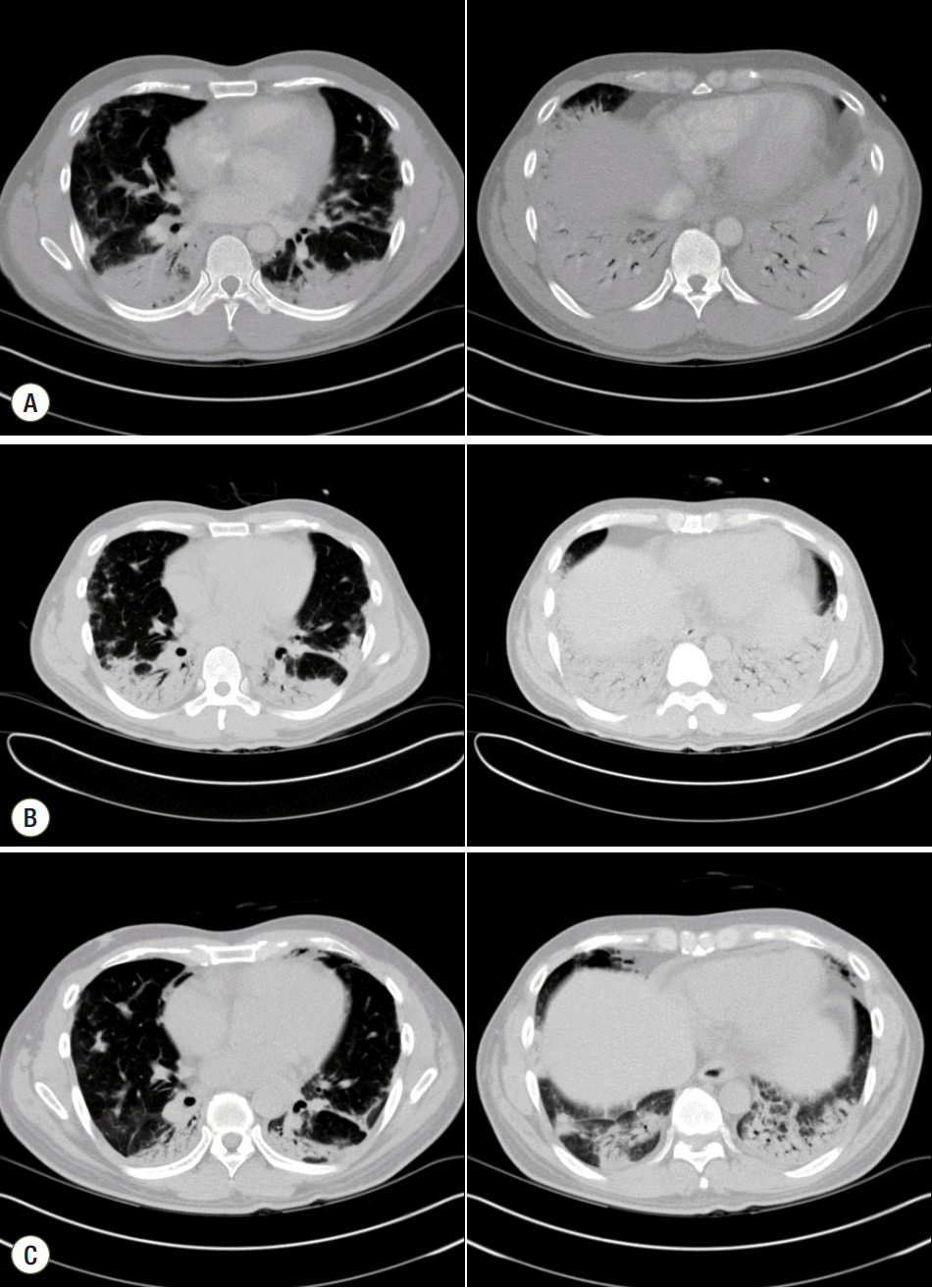 | Fig. 2.Chest computed tomography (CT). Chest CT at admission, showing peripheral consolidations in both lungs with ground-glass opacity mainly in the dependent portion (A). Chest CT on day 15 of hospitalization, showing worsening of the peripheral consolidation in both lower lungs, as well as bronchovascular thickening (B). Chest CT on day 31 of hospitalization, showing improvement of the organizing pneumonia (C). The severity of consolidation that had been dominant in the peripheral and dependent portions of both lungs was reduced.
|
On the basis of these findings, ILD and acute respiratory distress syndrome combined with dermatomyositis were diagnosed. The patient was treated with 4th generation cephalosporin and macrolide antibiotics, together with methylprednisolone 125 mg, and was admitted to the intensive care unit. He was maintained at FiO2 50% with a partial rebreathing mask. He remained in intensive care for 6 days. After his vital signs had stabilized and there was no worsening he was transferred to a general ward.
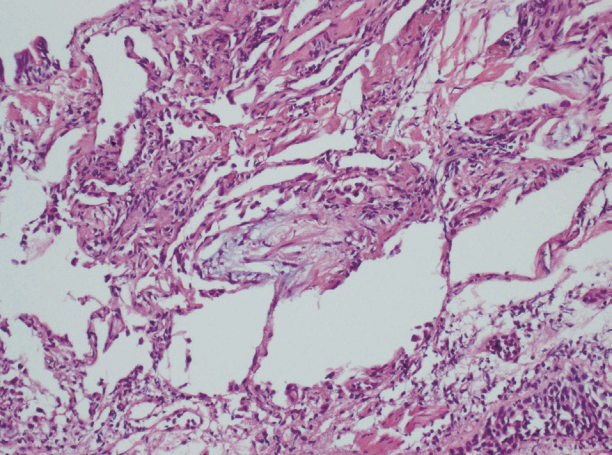
He continued on methylprednisolone 125 mg for the first week of hospitalization. On day 8, this dose was increased to 250 mg because there was no improvement in chest X-ray or dyspnea; the macrolide was stopped but the cephalosporin was continued. On day 14 of hospitalization, chest X-ray showed no improvement and he was maintained on an oxygen level FiO2 50%. A PaO2/FiO2 of 163 mmHg was confirmed. Antibiotic treatment was stopped because there was no clear sign of infection; methylprednisolone was reduced from 250 mg to 125 mg. Comparison of the chest CT on day 16 of hospitalization with the CT on day of admission showed a worsening without effective response to the high-dose steroid treatment. (Fig. 2) The patient underwent bronchoscopy and biopsy on day 17. The biopsy showed lymphoplasmacytic infiltration in the interstitium and a young fibroblastic nodule (Fig. 4); however, there was no sign of infection in the bronchial washing. Beginning on day 18, the patient was administered methylprednisolone 250 mg and oxygen at FiO2 40%. Although steroids were given at an appropriate dose, the COP did not improve. On days 22 and 23 he was given cyclophosphamide 500 mg/m2, but the chest X-ray on day 23 showed no improvement of the pulmonary infiltration (Fig. 1).
On day 23, treatment with i.v. immunoglobulin was started at 2 g/kg for 5 days. On day 25 the chest X-ray showed improvement in pulmonary infiltration on both lower lobes and the patient was given oxygen at FiO2 35% and methylprednisolone 125 mg. On day 27 he inhaled oxygen through a nasal cannula. The chest X-ray showed pneumomediastinum; however, the patient had no clinical symptoms and began to improve on day 29. Chest X-ray (Fig. 1) and CT (Fig. 2) on day 31 indicated improvement in pulmonary consolidation and pneumomediastinum. On day 32 the dose of methylprednisolone was reduced to 90 mg, and on day 35 the dose was further reduced to 30 mg twice a day. The patient was maintained on this dose until discharged from the hospital. Prednisolone 30 mg twice a day was prescribed at the time of discharge. After discharge, the patient was hospitalized on two more occasions. Cyclophosphamide 500 mg/m2 was administered and his progress is now being observed in the outpatient clinic. He is currently taking hydroxychloroquine, azathioprine, and prednisolone 15 mg/day.
Go to : 
Discussion
In ILD caused by connective tissue disease, damage to the endothelium and alveolar epithelium occurs through microvascular injury resulting from autoantibody attack on connective tissue. Various forms of ILD occur in connective tissue diseases such as systemic sclerosis, rheumatoid arthritis, polymyositis, dermatomyositis, Sjögren’s syndrome, systemic lupus erythematosus, and mixed connective tissue disease.[6]
Dermatomyositis is very rare even among rheumatic diseases, with an annual incidence of only 2.2 to 7.7 cases per million population. The prevalence of all types of ILD with dermatomyositis ranges from 5% to 30% depending on diagnostic method.[7] Usually its form is chronic progressive ILD form represented as nonspecific interstitial pneumonia, usual interstitial pneumonia or COP. The incidence of acute respiratory failure is approximately 5% or below.[8] Cases with the COP pattern have a survival rate between 50% and 80%, cases of usual interstitial pneumonia approximately 33. However COP with a more aggressive clinical course and unresponsiveness to steroid is associated with high mortality.[9,10]
In ILD, chest X-ray is a useful method of medical image screening but has several shortcomings for confirming initial-phase ILD. Hence high-resolution CT is widely considered the standard method for initial testing of patients suspected of ILD; the image may show, for example, a reticular pattern or ground-glass opacity. Bronchoalveolar lavage is not specific for myositis in diagnosing the involvement of lung parenchyma; however it is useful in differential diagnosis of other causes, such as infection, drug-induced reaction, or malignancy. Moreover, it can be helpful for determining disease activity or prognosis. As in idiopathic pulmonary fibrosis, cases in which the eosinophil count is increased or there is a finding of neutrophil-dominant alveolitis imply progressive disease or a poor prognosis. In that situation, high-resolution CT for diagnosis and bronchoalveolar lavage to rule out infection are used.[11]
Immunosuppressive agents including steroids are used to treat ILD combined with dermatomyositis. Prednisone 1 mg/kg is used for initial and maintenance treatment and is tapered in accordance with the symptoms of patients.[12,13] When ILD is severe or progressive, secondary immunosuppressive agents such as azathioprine, mycophenolate, cyclophosphamide, or methotrexate are administered, which also permits reduction of the steroid dose. Moreover, in patients whose ILD is progressing despite the use of steroid and cyclophosphamide, it is possible to add a third immunosuppressive agent, a calcineurin inhibitor.[14] Rituximab has been found effective in severe progressive ILD not responsive to existing immunosuppressive agents.[2-4]
Although there is little is known of how immunoglobulin acts in patients with ILD, i.v. immunoglobulin is still widely viewed as an effective and safe treatment for polymyositis or dermatomyositis. In one study, approximately 70% of patients were reported to show an improvement in myopathy, skin lesions, and esophageal dysfunction;[15] in another study where immunoglobulin was given to five patients with refractory ILD that had not responded to other treatments, two (40%) showed improvement in the PaO2/FiO2 ratio. [5] In a third report, a 64-year-old patient who suffered an adverse effect from steroid and secondary immunosuppressive agents was treated successfully with i.v. immunoglobulin,[16] and in a further report, immunoglobulin treatment in progressive ILD resulted in positive progress.[17]
At present, the treatment for ILD combined with dermatomyositis is mainly steroid and secondary immunosuppressive agents. There is no clear recommendation for use of intravenous immunoglobulin and no convincing mechanism of action has been presented. Nonetheless, some studies have reported that immunoglobulin is effective in treatment of dermatomyositis and ILD. In the present case, i.v. immunoglobulin was used successfully to treat ILD unresponsive to steroid and immunosuppressive agents.
Go to :
 XML Download
XML Download