

 PDF
PDF Citation
Citation Print
Print



Introduction
Cardiovascular disease (CVD) is the leading cause of mortality worldwide, accounting for 17.9 million deaths each year [1]. The main underlying cause is atherosclerosis. Compromised stability of atherosclerotic plaques results in thrombus formation, and, consequently, stroke or myocardial infarction (MI) depending on the affected territory. Characteristics of a classically rupture-prone or vulnerable plaque include a thin fibrous cap, abundance of lipids, a prominent necrotic core and infiltration by immune cells, such as macrophages and T cells [2], while eroded lesions are less inflamed and rich in proliferating smooth muscle cells (SMCs), proteoglycans and hyaluronan, and neovascularization [3]. Ideally, ameliorating atherosclerosis involves two steps: (1) finding a reliable method to identify individuals with vulnerable plaques to start treatment in time to prevent ruptures—and subsequently symptoms, and (2) identifying key players driving destabilization in rupture-prone plaques. Addressing both steps, this study not only evaluates the CD40/CD40L signalling dyad as a potential CVD marker but also explores its biological association with a vulnerable plaque phenotype.
CD40 and CD40 ligand (CD40L) are costimulatory molecules and members of the tumor necrosis factor (receptor) superfamily (TNF(R)SF) [4]. CD40 is expressed on a variety of immune and non-immune cells, including B cells, monocytes/macrophages, dendritic cells, SMCs and endothelial cells. CD40L is mainly expressed by activated T cells and platelets, but expression has also been described on B cells, as well as on endothelial, epithelial, and SMCs [5] CD40 signalling is propagated via adaptor proteins—the TNF receptor-associated factors [6] —and modulates a multitude of immunological responses [5]. Atherosclerotic mouse models (apolipoprotein E [ApoE-/-], low-density lipoprotein receptor [LDLr-/-]) with disrupted CD40 signalling develop lesions of reduced size with a more stable plaque profile, an effect achieved by CD40/CD40L knock out models, as well as by small molecule inhibitors and antibodies inhibiting the CD40 signalling pathway [6-9].
While not much is known about the specific actions of CD40 in human CVD, an association for CD40L in CVD has been suggested. Increased levels of platelet CD40L have been reported in patients with hypercholesterolemia [10] and plasma levels of soluble CD40L (sCD40L) were increased in patients with acute coronary syndromes [11]. Furthermore, clinical evidence for involvement of sCD40L in CVD include association with cardiovascular (CV) events in coronary artery disease [11], recurrent cardiac events and future events in healthy women [12], carotid intima-media thickness (IMT) and hypertension [13], and via correlations with other biomarkers of inflammation [14]. A number of studies, however, have found no associations between plasma sCD40L and CVD [15,16] or long-term CV (or all-cause) mortality [17]. Further clarification is thus essential to determine the value of sCD40L as a marker of CVD, and to investigate whether sCD40 may be a useful complement or replacement biomarker.
To address the potential of sCD40 and sCD40L as markers of CVD, we have analysed plasma and carotid artery plaque levels of CD40/CD40L for associations with CV events, as well as carotid IMT and plaque vulnerability markers in two large human cohorts.
Methods
Extended methods are provided in the Supplementary Methods.
Study cohorts and samples
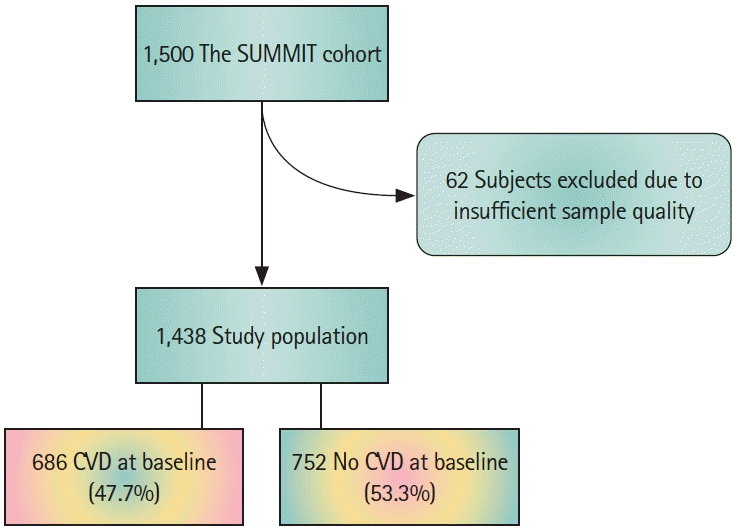
Plasma samples were included from 1,438 subjects (Figure 1) of the SUrrogate markers for Micro- and Macro-vascular hard endpoints for Innovative diabetes Tools (SUMMIT) study [18] population, recruited between December 2010 and April 2013 from existing population cohorts and hospital registries at the university hospitals in Malmö (Sweden), Pisa (Italy), Dundee and Exeter (UK).
Carotid plaques (n=198) were collected from patients during carotid endarterectomies at the Vascular Department of Skane University Hospital (Malmö, Sweden) between 2005 and 2010 (The Carotid Plaque Imaging Project [CPIP]) [19]. Immediately during surgery, removed plaques were snap-frozen in liquid nitrogen. Histological analyses were performed on portions (1 mm thick) cut from the most stenotic plaque region and embedded in optimal cutting medium (Sakura Finetek Europe BV, Tokyo, Japan). Plaque homogenates were prepared from the rest of the plaques and used for all other plaque component quantifications. Clinical characteristics of the included patients are summarized in Supplementary Table 1. The studies were approved by the local ethical review boards and were carried out in accordance with the principles of the Declaration of Helsinki. There was no overlap between subjects of the two study cohorts.
Statistics
Shapiro-Wilk and D’Agostino-Pearson omnibus K2 tests were used to assess Gaussian distribution. Variables found to be normally distributed are shown as mean with standard deviation while non-normally distributed variables are shown as median with interquartile range (IQR). For analysis of plasma, plaque histology sections and homogenates, the Mann-Whitney U test was used to compare groups and Spearman’s rank correlation was used for continuous variables. For comparisons between categorical variables, the chi-square test was used. A logistic regression model was used to test for associations with CV events and mortality in the SUMMIT cohort. The model was adjusted for age, diabetes and prevalent ischemic stroke, acute myocardial infarction (AMI), and hypertension as covariates. A P-value of <0.05 was considered statistically significant. Statistical analysis was performed using SPSS version 26.0 (IBM Co., Armonk, NY, USA) and GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, CA, USA, www. graphpad.com). Adjustments for multiple comparisons were done when appropriate using the Holm-Šídák test.
Results
Plasma sCD40 levels are associated with stroke and acute myocardial infarction
To assess associations between sCD40, sCD40L and CVD, we analysed the plasma from 1,438 subjects from the SUMMIT cohort using Proximity Extension Assay (PEA). Comparatively high sCD40 levels were associated with prevalent ischemic stroke (P=3.0×10–5), MI (P=0.016), and hypertension (P=0.00021) (Table 1 and Supplementary Tables 2 and 3). The association with stroke remained after adjusting for age, diabetes, hypertension, and MI (odds ratio [OR], 1.5; 95% confidence interval [CI], 1.084 to 1.959; P=0.013) and the association with MI after adjusting for age, diabetes, hypertension, and stroke (OR, 1.2; 95% CI, 1.006 to 1.393; P=0.042).
Similar to sCD40, higher plasma sCD40L levels were associated with prevalent stroke (P=0.020). However, sCD40L levels were not associated with hypertension or prevalent AMI, and associations did not remain after adjusting for the aforementioned confounders (stroke: OR, 1.1; 95% CI, 0.976 to 1.304; P=0.103; and MI: OR, 1.0; 95% CI, 0.968 to 1.125; P=0.270).
Plasma sCD40 levels are associated with carotid plaque burden and arterial stiffness
Plaque burden in the carotid artery was assessed via ultrasound imaging at study inclusion and we searched for possible associations between sCD40 and sCD40L and plaque burden. A positive Spearman’s rank correlation was found between both plasma sCD40 and sCD40L levels and total plaque area (r=0.355, P<1×10–16 and r=0.409, P<1×10–16, respectively) (Table 2). The association between sCD40 and sCD40L and carotid plaque burden was also reflected by positive correlations with overall number of plaques, measurements of plaque length, height and area, as well as with lumen diameter reduction (Table 2).
Arterial stiffness—a well-known factor associated with atherosclerosis progression [20]—was studied by pulse wave velocity at study inclusion. Plasma levels of sCD40, but not sCD40L, correlated with carotid arterial stiffness (r=0.247, P<1×10–16) (Table 2). Furthermore, sCD40 plasma levels measured at study inclusion showed a positive correlation with both mean and maximum IMT in the right and left common carotid artery (CCA), as well as in in the plaque bulb regions (Table 3). Association was also found between plasma sCD40L and mean IMT in the bulb region of the right carotid artery (Table 3).
Plasma sCD40L levels are associated with carotid artery plaque progression and sCD40 levels with future cardiovascular events
To elucidate whether plasma sCD40 and sCD40L levels predicted carotid atherosclerosis progress, in addition to correlating with plaque burden at study inclusion, we compared the initial IMT measurements calculated via ultrasound imaging with IMT measurements taken after 3 years. Plasma sCD40L, but not sCD40, levels were associated with change—specifically plaque growth—in maximum IMT in the CCA and mean and maximum IMT in the bulb region of the plaques (Supplementary Table 4).
As sCD40L levels correlated with carotid plaque progression, we then investigated the potential for plasma sCD40 and sCD40L levels to predict future CV events using logistic regression. Interestingly, the level of sCD40, but not sCD40L, in plasma was associated with the occurrence of fatal or non-fatal CV events during the 3 years follow-up period (OR, 1.3; 95% CI, 1.042 to 1.625) after adjusting for age, diabetes, hypertension as well as prevalent AMI and stroke at baseline (Table 3). Neither sCD40 nor sCD40L was associated with mortality (CV or all-cause). Considering sCD40 (with confounders age, diabetes, hypertension as well as prevalent MI and stroke at baseline) in a multiple logistic regression, the area under the curve achieved was 0.629 (95% CI, 0.582 to 0.677), with sensitivities and specificities for varying cut-offs of the predicted values (10% to 90% quantiles) presented in Supplementary Table 5.
CD40 and CD40L have similar expression patterns in human carotid plaques
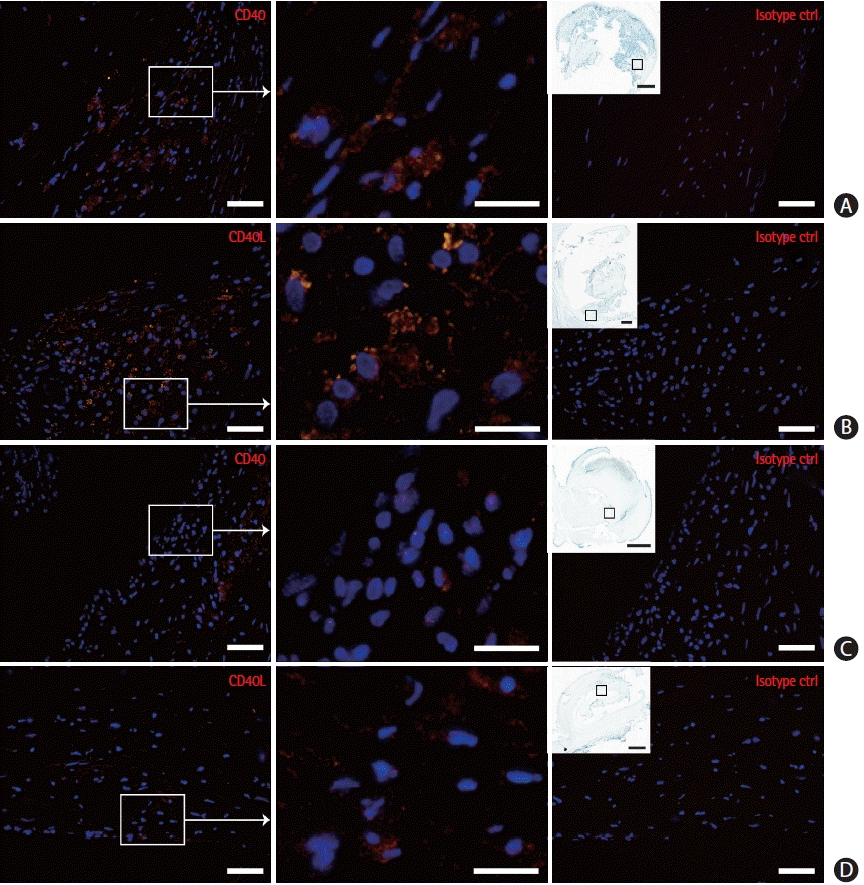
To further explore the presence of CD40 and CD40L in atherosclerosis we examined their expression pattern in carotid endarterectomy plaques from the CPIP cohort (Figure 2). Immunofluorescence staining was performed on sections obtained from the most stenotic plaque region. CD40 and CD40L expression was observed in several plaque parts, in particular in the shoulder and cap regions. Immunoreactivity was also found—though far less prominent—in the base of the plaque.
sCD40 is associated with a vulnerable plaque phenotype
To further explore the presence of CD40 and CD40L in atherosclerosis we examined their expression in homogenates from 198 carotid endarterectomy plaques of the CPIP cohort (here comprised of both the soluble and cell associated isoforms). Disrupted CD40 signalling in a murine atherosclerosis model results in increased stabilisation characterised by reductions in lipid core size and inflammatory cell presence accompanied by increased collagen content [6-9]. We therefore investigated whether intra-plaque levels of these molecules are similarly associated with a vulnerable plaque profile in human lesions. Indeed, we found sCD40 plaque levels measured in plaque homogenates of the CPIP cohort to correlate with plaque components collectively indicating a vulnerable plaque phenotype (Table 4). Accordingly, there was a correlation between sCD40 and lipid content (visualized on tissue sections by Oil Red O, r=0.199, P=0.012; and oxidized low density lipoprotein [oxLDL] measured in plaque homogenate, r=0.236, P=0.004), whereas a negative correlation was found with calcium (von Kossa, r= –0.208, P=0.012). The level of sCD40L in plaque showed an overall association with a vulnerable plaque phenotype, albeit weaker than that for sCD40 levels (Table 4). sCD40L levels correlated with the plaque content of lipids (oxLDL measured in plaque homogenate, r=0.259, P=0.0037), while there was an inverse correlation with plaque levels of calcium (von Kossa, r=–0.268, P=0.00034). Finally, for further plaque profile evaluation a vulnerability index was calculated, which was also found to correlate with both sCD40 (r=0.178, P=0.029) and sCD40L (r=0.180, P=0.041) plaque levels.
CD40 ligation has been previously well-described to induce pro-inflammatory cytokine and chemokines, including interleukin (IL)-1β, IL-6, IL-8, IL-12, TNF-α, and chemokine (C-C motif) ligand 5 (CCL5) in vitro [21-23]. Concordantly, sCD40 levels correlated with both IL-12p70 and CCL2 measured in plaque homogenate, as well as with several other inflammatory cytokines and chemokines, including IL-6, TNF-α, CCL4, and C-X-C motif chemokine ligand 1 (CXCL1) (Table 4). Correlation was also found with platelet-derived growth factor (PDGF). Similarly, sCD40L levels correlated with the inflammatory cytokines and chemokines IL-12p70, CCL2, IL-6, TNF-α, CCL4, CCL5, CXCL1 as well as with PDGF (Table 4).
sCD40 plaque levels are associated with features of extracellular matrix turnover
The reported increase in fibrous cap thickness and collagen content in plaques from CD40-deficient mice has been proposed to result from the accompanying decreased plaque expression and activity of matrix metalloproteinase (MMP)-2 and -9 together with increased tissue inhibitor of metalloproteinase (TIMP)-1 expression, and has thus prompted the suggestion that CD40 signalling destabilizes plaques by promoting extracellular matrix (ECM) degradation and, as a result, fibrous cap thinning [6]. Therefore, in this study we searched for similar associations in human samples. Plaque levels of CD40 correlated with plaque content of MMP-1, -2, -9, and -10, while sCD40L levels correlated with only MMP-10 (Supplementary Table 6) Furthermore, both sCD40 and sCD40L levels correlated with TIMP-1 and TIMP-2. Thus, sCD40 (and CD40L) were indeed associated with plaque ECM turnover. Of note, correlations were also found between both sCD40 and sCD40L levels and content of intact elastin and collagen (type I–V) measured in plaque homogenate.
Plaque sCD40 and sCD40L levels are not associated with pre-operative cerebrovascular symptoms
As sCD40 and sCD40L plasma levels were higher in subjects who had suffered a stroke in the SUMMIT cohort, we next analysed whether sCD40 and/or sCD40L levels are similarly elevated in the plaques from individuals after a cerebrovascular event (median time between symptom and surgery was 14 days [IQR, 7 to 22]; the CPIP cohort). Levels of CD40 and CD40L measured in plaque strongly correlate with each other (r=0.613, P<0.0001) but neither sCD40, nor sCD40L levels correlated with the time between symptoms exhibited by the patients and the time of endarterectomy (r=0.04, P=0.688 and r=0.063, P=0.523, respectively). Also, plaque levels of sCD40 and sCD40L were similar in plaques from asymptomatic and symptomatic patients (n=102 and n=96, respectively).
Discussion
With this study, we set out to assess the value of sCD40 as a possible marker of CVD and investigate the biological associations that support a role for CD40 in atherogenesis. Our findings that sCD40 levels in plasma are associated with carotid plaque burden, while levels in carotid plaques are associated with a vulnerable plaque phenotype, strengthens the potential for sCD40 as marker for and mediator of CVD.
In contrast to the paucity of data in the literature for sCD40 as a marker for CVD, sCD40L has been previously widely linked to CVD via increased levels in patients with hypercholesterolemia [10] and acute coronary syndromes [11,24]. Upregulated platelet surface expression of CD40L was also reported to correlate with worse clinical outcome after stroke [25] and plasma sCD40L levels were higher in patients with carotid artery lesions that have intra-plaque lipid as detected by high-resolution magnetic resonance imaging [26]. Furthermore, Balla et al. [27] identified serum sCD40L levels as a risk factor for increased IMT in subjects with white-coat hypertension. Our current results are in line with these studies.
Much less is known about the possible role of sCD40 as a marker for CVD. Though sCD40 appears to be actively produced under physiological conditions (as suggested by comparatively large amounts of sCD40 found in urine of healthy individuals) [28], elevated plasma levels of sCD40 has nonetheless been described in pathologies such as systemic lupus erythematosus [29], slow coronary flow phenomenon [30] and early Alzheimer’s disease [31]. In the SUMMIT cohort, we found higher plasma sCD40 levels in individuals that had suffered a prior ischemic stroke or MI or were hypertensive. Plasma sCD40 levels also correlated with carotid plaque burden and were associated with future CV events over a follow-up period of 3 years. We therefore propose sCD40 as a potential marker for the presence and severity of CVD.
Efficient targeting of inflammatory processes can successfully stabilize vulnerable plaques in mice [8,9] but this approach is not yet utilized in human disease beyond clinical trials [32] due to detrimental side effects. In atherosclerotic mouse models disrupted CD40 signalling—after blockade with antagonistic antibodies, small molecule inhibitors and in CD40- and CD40L-deficient mice—consistently results in plaques of reduced size and, importantly, a stabilized phenotype characterized by reduced lipid core content, macrophage and T-lymphocyte content accompanied by increased fibrous cap thickness, and relative collagen and SMC cell content [6-9]. Previous studies exploring CD40 signalling in relation to severity of inflammatory diseases in humans have mainly focused on systemic levels of the signalling dyad, in particular levels of sCD40L and CD40L. This is the first study to also investigate associations between carotid intra-plaque CD40/CD40L content and plaque phenotype and disease progression. We found intra-plaque sCD40 (and to a certain extent also sCD40L) levels to be associated with a plaque profile rich in lipids, MMPs and pro-inflammatory cytokines and chemokines: destabilising elements corresponding to those affected by blocking of the CD40/CD40L axis in mice [6-9]. This link between human disease and mouse model strengthens the merit of translating therapeutic targeting of CD40 from the academic setting one step closer to the clinical application.
Neither CD40 nor CD40L was associated with time between cerebrovascular symptoms and endarterectomy, suggesting they are not merely upregulated as part of the general healing response that occurs after plaque rupture. However, though sCD40 levels were associated with a less stable carotid plaque phenotype, plaque levels were nonetheless similar in plaques from symptomatic and asymptomatic patients. This inconsistency may, at least in part, be explained by the accompanying association with components of the ECM turnover machinery, leading to a net effect in CD40-rich plaques, where enough ECM synthesis of stable plaque elements balances out the more vulnerable elements, resulting in a plaque phenotype still stable enough not to rupture/erode and cause cerebrovascular symptoms. This in turn may indicate a local protective element in CD40-CD40L signalling, in addition to its well-described actions promoting the inflammatory process. We hypothesize that this may demonstrate the contrasting results of CD40 and CD40L interactions with (and in) different cell types of varying functions, such as macrophages, T cells, and SMCs in a model where CD40-signalling simultaneously for example promotes inflammation via macrophages and ECM synthesis via SMCs, even if the phenotypes of these cells have also been widely discussed [33]. The therapeutic angle described by Seijkens et al. [9] of targeting CD40 signalling blockade specifically to macrophages (via incorporation into reconstituted high-density lipoproteins nanoparticles) therefore also emerges as an especially relevant approach to achieve plaque stabilisation.
Also interesting from a therapeutic standpoint is the study by Fernandez et al. [34], in which single cell RNA sequencing was compared in carotid plaques from patients who had or had not experienced cerebrovascular symptoms. Unexpectedly, the most upregulated pathways in T cells and macrophages from plaques from symptomatic patients were those of repair, while pro-inflammatory pathways were elevated in plaques from asymptomatic patients. Repair pathways are thus deduced to be induced by plaque rupture, and therefore represent a less striking presence in plaques whose integrity is more intact. Consequently, inflammatory activity weighs heavier in plaques from asymptomatic than from symptomatic individuals. This notion confirms the significance of our report that sCD40 is associated with vulnerable plaque components such as lipid content and pro-inflammatory cytokines, as well as with overall plaque burden, regardless of whether symptoms have manifested or not. Furthermore, the great success in ameliorating size and vulnerability of murine lesions (that are, per definition, pre-rupture) by blocking CD40 signalling affirms the value of targeting inflammatory responses in non-ruptured lesions.
As limitations, it must be noted that correlations do not indicate causality, and we cannot rule out that the reported associations may also result from an upregulation of sCD40 and sCD40L stimulated by an upstream signalling cascade or events. It is also important to bear in mind that in the plaque homogenates derived from the CPIP cohort it is not possible to distinguish between soluble and cell-bound CD40 and CD40L. The analysis thus shows the net effect of both isoforms, even if the effects of the individual isoforms may differ. Moreover, though it is impossible to be certain of the exact type of ischemic stroke, as subjects with atrial fibrillation were excluded in both cohorts, and—in the CPIP cohort—no signs of lacunar infarctions were found (assessed by brain computed tomography), the probability of the underlying mechanisms to be atrial fibrillation and small vessel disease rather than atherosclerosis can be considered low. Unfortunately, the presence of cardiac valvular disease is impossible to exclude as not all patients underwent echocardiography. Finally, as we in both cohorts measured sCD40L in plasma to evaluate its potential as a non-cell associated marker, platelet expression of CD40L—previously reported e.g., by Cha et al. [35] to be higher in subjects with atherosclerotic ischemic stroke than with asymptomatic carotid stenosis—has not been taken into account.
Conclusions
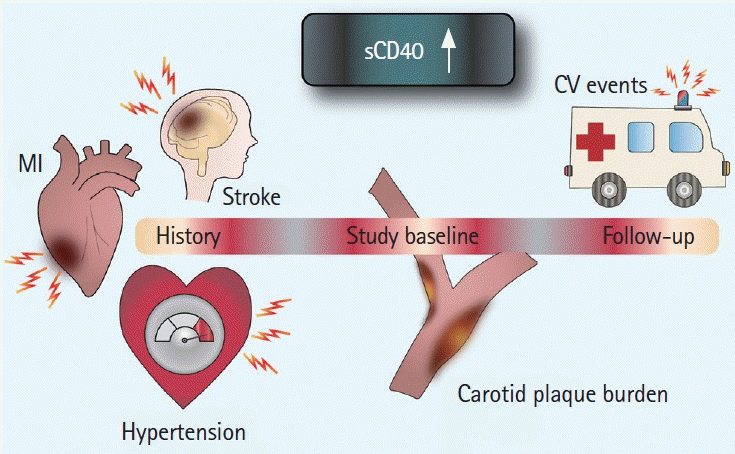
In the present study we report for the first time elevated plasma sCD40 levels in individuals with a prior ischemic stroke or MI, as well as an association with arterial stiffness, carotid plaque burden, and IMT, and, finally, an association with increased risk of future CV events (Figure 3). Plasma levels of sCD40L are confirmed to not only be elevated in subjects after stroke, but also to correlate with future plaque progression in the form of IMT increase. We also present a novel association between intra-plaque levels of CD40 and CD40L and a vulnerable plaque phenotype.
 XML Download
XML Download