

 ePub
ePub Citation
Citation Print
Print


A fixed pupillary light reflex after major surgery may indicate a life-threatening neurological condition. In a patient with end-stage liver disease and accompanying severe coagulopathy, intraoperative spontaneous cerebral hemorrhage should be considered when the immediate postoperative pupillary light reflex is not normal after surgery. In the case of acute massive intracranial hemorrhage, immediate neurological workup and treatment is required. However, benign conditions such as peripheral somatic and autonomic neuropathy or Holmes-Adie syndrome (HAS) are also associated with fixed pupillary light reflexes.[1,2] We report a case of a fixed pupillary light reflex after liver transplantation in a patient diagnosed with peripheral autonomic neuropathy.
Case Report
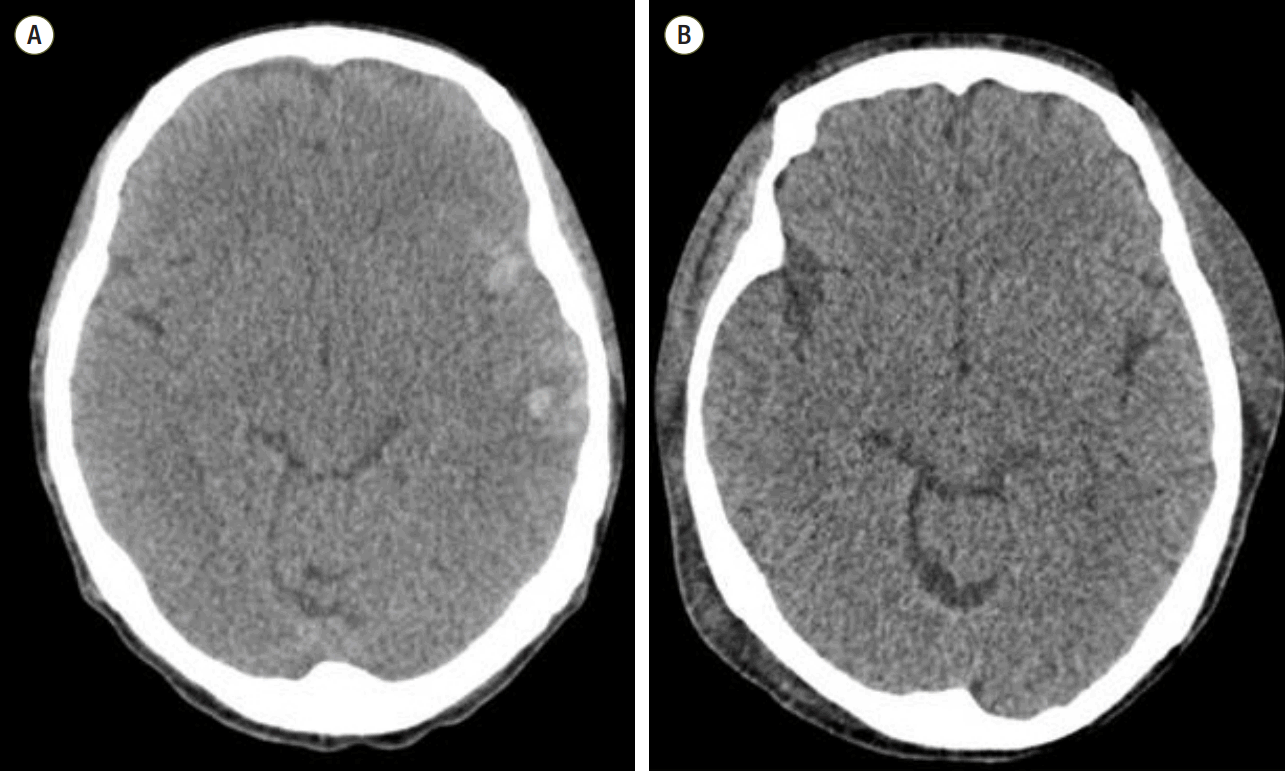
A 46-year-old female patient was admitted to the intensive care unit (ICU) after liver transplantation due to B-viral liver cirrhosis; the anesthetic time was about 13 hours. She had a history of diabetes mellitus (DM) for several years, which was inappropriately managed despite the use of insulin administration with 11.2% HbA1c before surgery. However, her intraoperative blood sugar level was strictly maintained from 100 to 200 mg/dL. One month before admission for liver transplantation, she had a falling accident resulting in a traumatic head injury with multifocal contusional intracranial and subarachnoid hemorrhage in the left cerebral hemisphere (Fig. 1A). However, at the time of the accident, there were no signs of neurologic deficits, including an abnormal pupillary light reflex. The cerebral hemorrhage spontaneously resolved without surgical intervention. Since the accident, symptoms related to liver cirrhosis became aggravated, including a distended abdomen and abdominal discomfort. Abdominal computed tomography (CT) revealed large amounts of ascitic fluid and splenomegaly. In addition, it became difficult to control the ascites.
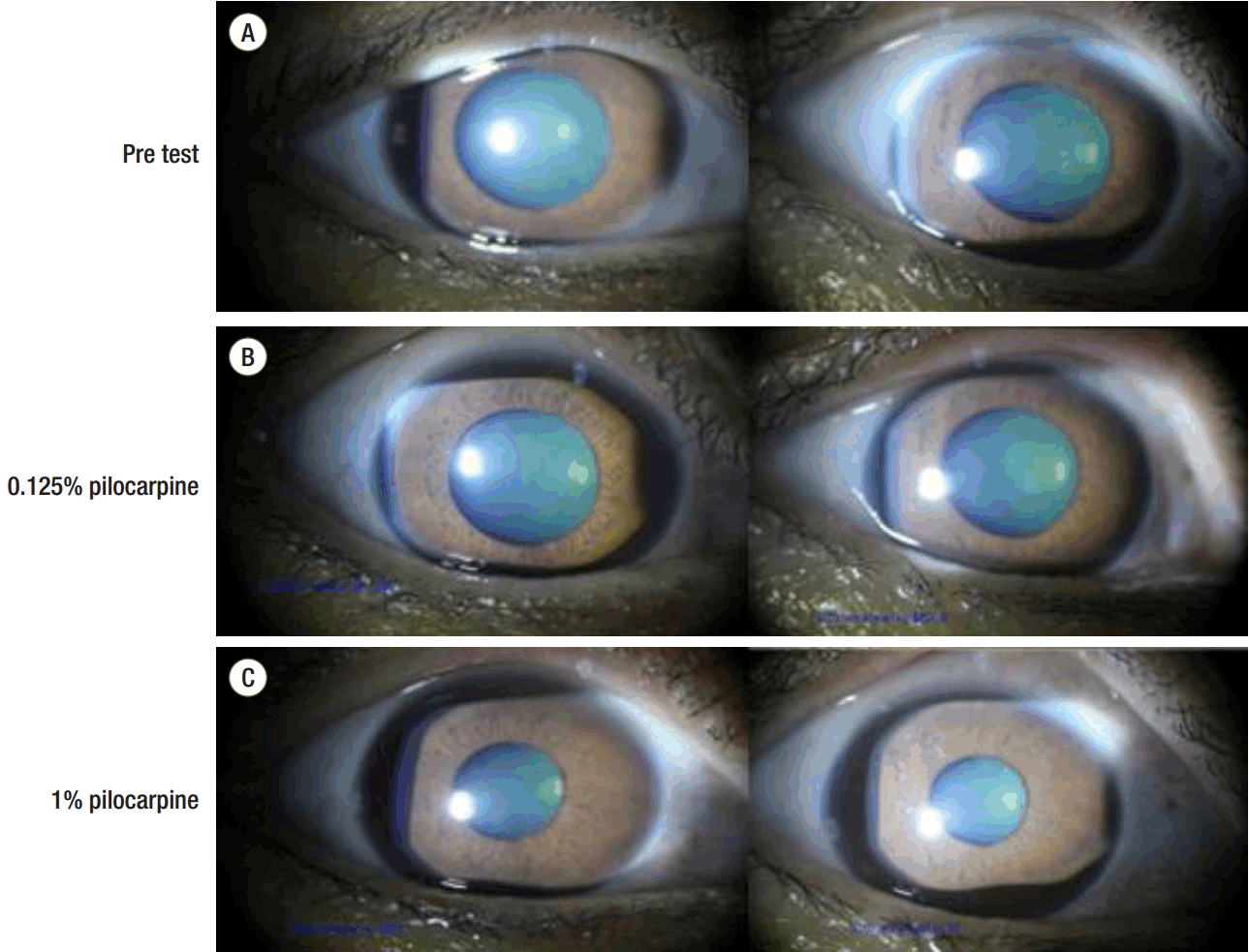
At the time of ICU arrival, the patient was intubated with isocoric pupils of about 5-mm diameter and had a hardly distinctive, sluggish pupillary reflex to light. We initiated sedation with propofol and remifentanil. About an hour later, no pupillary light reflex was observed, and both pupils were fixed at 5 mm. We immediately discontinued the drugs for sedation (propofol and remifentanil). A brain CT scan was also performed. Although there were no signs of a spaceoccupying lesion on the brain CT, the previously noted trauma-associated hemorrhage in the left cerebral hemisphere had decreased in density (Fig. 1B). She was arousable 1.5 hours after drug discontinuation. Twelve hours after ICU arrival, she was extubated successfully after the performance of a spontaneous awakening trial and spontaneous breathing trial. However, her pupils remained unchanged. Upon neurological examination, deep tendon reflexes including the knee jerk and ankle jerk were absent. Other signs were within normal ranges. Two months later, there was no change in both pupils. Instillation of a dilute solution of pilocarpine (0.125%) in both eyes did not constrict the pupils. However, the pupils were constricted after 1% pilocarpine instillation, and there was no sector palsy or light anisocoria (Fig. 2). There were also no other neurologic deficits, excluding the absence of deep tendon reflexes. With this ophthalmologic evaluation, we could rule out the initially suspected HAS because in a patient afflicted with HAS, his or her pupils would have constricted with the instillation of dilute (0.125%) pilocarpine. Pupillary constriction to 1% pilocarpine implies a third nerve lesion. Thus, we strongly suspected generalized peripheral or autonomic neuropathy, which may be observed in end-stage liver disease or DM.
Discussion
Severe chronic liver disease is associated with the presence of peripheral somatic and autonomic neuropathy.[3-5] Although 76% of patients with end-stage liver disease present with abnormal autonomic function,[6] there have been only a limited number of reports about such cases.[6] Clinical peripheral neuropathy is reported with various incidences. Approximately 19% to 91% of patients with end-stage liver disease develop peripheral neuropathy.[3-5]
It is also known that all patterns of neuropathy can occur in patients with DM.[7] Diabetic peripheral neuropathy (DPN) is a common complication affecting 13% to 58% of patients with DM.[8,9] Generalized DPNs can be classified into two major groups (typical and atypical).[10] Typical DPN is a chronic, symmetrical, length-dependent sensorimotor polyneuropathy,[11] and certain diagnostic symptoms and signs including decreased sensation, neuropathic symptoms, and abnormal nerve conduction are required for diagnosis. Atypical DPN is not as well-characterized and studied as typical DPN. It appears to be an intercurrent and monophasic or fluctuating disorder, developing at any time during the course of a patient’s DM. Further surveys and studies are required for the classification of atypical DPN.[7]
Another probable diagnosis suspected in this case was HAS. HAS is characterized by initially monolateral and then bilateral enlargement of the pupil with a delayed response to the near vision effort. Typical pupil findings include attenuation of the light response, sector palsy, and a tonic near response. Deep tendon reflexes are absent. The condition was first reported in 1931,[12] and it is more common in women with a peak incidence in the third decade of life.[2,13,14] The condition typically affects one eye, (occasionally both) and has no serious implications.[15] Although the pathophysiology of HAS remains unclear, changes in the ciliary and spinal ganglia have been observed in post-mortem studies.[14,16] The tonic pupil is the result of damage to the parasympathetic ciliary ganglion by partial denervation of the sphincter muscle.[14,17] Denervation makes the iris sphincter hypersensitive to acetylcholine, and this hypersensitivity to acetylcholine can be used as a clinical test. Instillation of dilute pilocarpine (0.125%) solution is followed by substantial constriction of the affected pupil in HAS,[2,14,18] and this response to dilute pilocarpine solution is a diagnostic key for HAS. HAS can also be detected in a symptomatic form during the disease course of the nervous system, most frequently in polyneuropathies.[2]
In this case, both eyes showed absence of the pupillary light reflex and fixed pupil diameters at 5 mm. An urgent brain CT scan was performed and revealed no newly developed abnormal findings with the exception of decreased density of the previously noted trauma-associated hemorrhage.
Both pupils were constricted after the instillation of 1% pilocarpine with no response to 0.125% pilocarpine. This finding does not imply HAS, but more likely contributed to a third nerve lesion because hypersensitivity of the iris sphincter to a low concentration of pilocarpine is observed in HAS. Thus, we could rule out the initially suspected HAS.
Diagnosis of HAS based on a unilateral tonic pupil with areflexia is very straightforward.[19] However, as shown in this case, although bilateral tonic pupils with areflexia may be apparent in HAS, this sign may also be observed in patients with generalized peripheral autonomic neuropathy. [20] Nevertheless, upon ophthalmologic evaluation, there was no definite sector palsy of the pupil and no response to dilute (0.125%) pilocarpine. These helpful additional findings guided us to make a differential diagnosis. If both sector palsy and light anisocoria are absent, generalized neuropathy must be considered (specificity of 90%).[1] This patient had neither sector palsy nor light anisocoria and had no response to 0.125% pilocarpine. Thus, we diagnosed generalized neuropathy involving the autonomic nerves.
It is important to distinguish HAS from generalized neuropathy because HAS has a more favorable and benign prognosis than generalized neuropathy, which is probably induced by end-stage liver disease or DM. Absence of the pupillary light reflex itself is more important because it can indicate a very urgent situation. Such an absent light reflex is suggestive of space-occupying lesions that involve the midbrain and may require immediate surgical or nonsurgical intervention. We also performed a diagnostic workup including an urgent brain CT scan to evaluate the possible causes of the sudden presence of fixed pupillary reflexes.
Because no preoperative neurologic physical examinations including the pupillary light reflex had been performed, we had no choice but to hasten the neurologic evaluations. Preoperative neurologic examinations are often omitted if there is no specific neurologic history, but they are very useful in postoperative situations, as in this case. If we had additional information on the abnormal pupils of the patient before the ICU admission, we could manage the situation and avoid fluster.
Conclusively, peripheral neuropathies observed frequently in patients with end-stage liver disease or DM can mimic neurologic emergencies with presentation of fixed pupillary light reflexes after liver transplantation. We suggest that preoperative neurologic examinations including a cranial nerve testing, sensory and motor examination, reflex testing, coordination testing, and gait testing should be performed in every patient with end-stage liver disease or DM before liver transplantation.
 XML Download
XML Download