

 PDF
PDF Citation
Citation Print
Print


Introduction
Hepatocellular carcinoma (HCC) is the most common type of liver cancer. In the last decade, it has become one of the most frequently occurring tumors worldwide, and it is the third highest cause of cancer-related death globally [1]. In Korea, HCC remains one of most common causes of cancer-related mortality, although the incidence rate of liver cancer has decreased since 1999 [2].
For patients with early-stage localized disease, surgical resection, liver transplantation, and ablative therapy are potentially curative treatment options [3]. Chemoembolization is the mainstay of treatment for patients with HCC confined to the liver with preserved liver function [3]. However, patients with HCC diagnosed at the advanced stages, or which has progressed after locoregional therapy, show poor prognosis. Sorafenib, a multikinase inhibitor, has been established as a standard first-line systemic chemotherapy for patients with advanced HCC based on the success of pivotal phase III trials [4,5]. After 10 years of failures in the development of systemic treatment for HCC, regorafenib, an oral multikinase inhibitor, recently demonstrated improved survival in patients who progressed on sorafenib in the randomized phase III RESORCE trial [6], and nivolumab, an anti–programmed-cell-death protein 1 (PD-1) inhibitor, showed promising activity in a non-comparative phase I/II Check-Mate 040 trial [7]. Despite these recent advances, the survival of patients with advanced HCC remains dismal and novel approaches to target and inhibit hepatocarcinogenesis are required to improve survival outcomes.
Signal transducer and activator of transcription 3 (STAT3) belongs to the STAT family of proteins, which have functions as both signal transducers and transcription factors [8]. The aberration of STAT3 appears to be inherent in its relationships with multiple types of hematological and solid cancers [8]. A link between STAT3 and HCC has been previously suggested, as enhanced production of interleukin 6 and tumor necrosis factor through the activation of STAT3 is critical for obesity-promoted HCC preclinical models [9,10], and the STAT3 signaling pathway is involved in multiple models of hepatocellular oncogenesis [11-13]. In addition, STAT3 inhibitors showed significant anti-cancer activity in preclinical HCC models [14]. This suggests that STAT3 is a promising target for the treatment of advanced HCC [15,16]. Although several STAT3 inhibitors were tested in advanced solid cancers, including HCC, no agent has been proven to be effective in a randomized trial [17-19].
OPB-111077 is an orally bioavailable STAT3 inhibitor which has shown preclinical efficacy in both in vitro and in vivo preclinical HCC models. Based on these rationales, we conducted a multicenter, open-label, dose-finding phase I study of OPB-111077 for patients with advanced HCC.
Materials and Methods
This study was a multicenter, open-label, non-comparative, dose escalating phase I study in patients with advanced HCC, to determine the dose-limiting toxicities (DLTs), maximal tolerated dose (MTD), and recommended dose (RD) of OPB-111077. The dosing escalation scheme included a continuous dosing (daily administration) and intermittent dosing (4-days on/3-days off administration) regimen. This study was conducted at five academic institutions in Korea.
1. Patients
Patients with HCC, confirmed by a pathological or noninvasive assessment according to the American Association for the Study of Liver Diseases criteria for patients with confirmed cirrhosis, were eligible for inclusion in this study if they met the following criteria: age between 20 and 75 years; Eastern Cooperative Oncology Group (ECOG) performance status of 0-1; extrahepatic metastasis or locally advanced disease not amenable to surgical resection or loco-regional therapies; Child-Pugh classification A or B (except 9); documented evidence of unresponsiveness to, intolerance to, or ineligibility for sorafenib; and estimated life expectancy of 12 weeks or greater.
2. Study design and treatment
A standard, multiple dose, 3+3 study design was used, and consisted of two dosing regimen cohorts: a continuous dosing regimen for once daily administration (5 cohorts with doses of 50 mg, 100 mg, 150 mg, 200 mg, and 250 mg, respectively) and an intermittent dosing regimen for 4-days on/3-days off administration (3 cohorts with doses of 300 mg, 500 mg, and 600 mg, respectively). Both dosing regimens consisted of a 24-day initial treatment cycle (cycle 1), and 21-day subsequent treatment cycles (cycle 2 onwards) (Fig. 1).
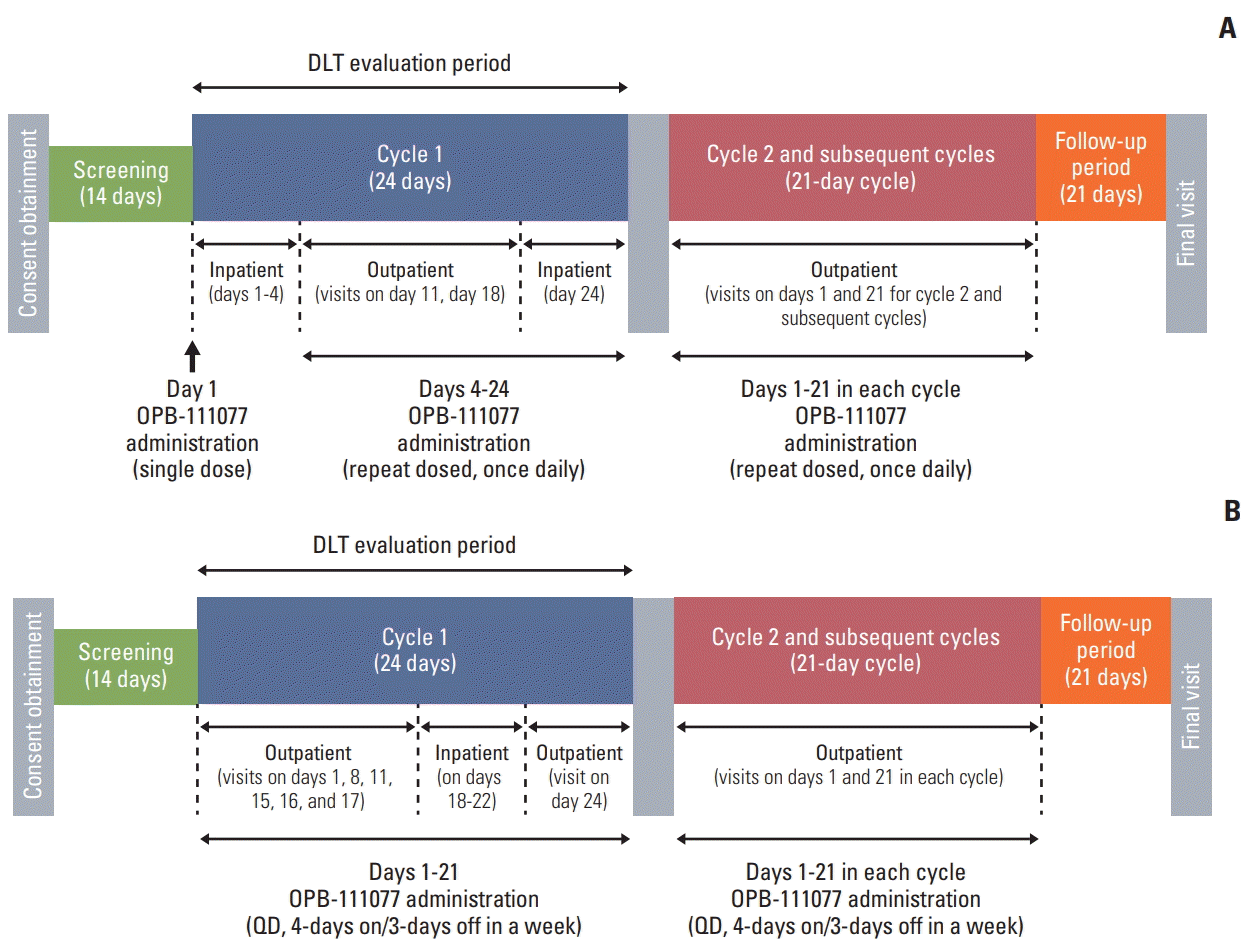
This study was originally designed to evaluate the continuous dosing regimen; however, intermittent dosing cohort was added subsequently based on the findings of a prior phase I study of OPB-111077 for patients with unselected advanced cancers [20] that all DLTs occurred at the 300 mg/day and 400 mg/day dose levels at approximately 1 week after the start of administration. Setting a washout period (3 days) was considered to ease the management of the enhancement of toxicity in the daily regimen of OPB-111077.
OPB-111077 was administered as a single oral dose at approximately the same time each morning. In cycle 1 of the continuous dosing regimen, patients received OPB-111077 on day 1, followed by a 2-day treatment-free interval for safety and pharmacokinetic (PK) evaluation. OPB-111077 administration resumed on day 4 and continued through day 24. In the intermittent dosing regimen cohort, patients received OPB-111077 for 4 days each week for 3 weeks (day 1 to 21), followed by a 3-day treatment-free period (days 22 to 24). Study treatment continued until patients experienced intolerable toxicity, the occurrence of disease progression, or withdrawal of consent. Cycle 2 and subsequent cycles consisted of 3 weeks (21 days) with no treatment-free intervals.
Doses were reduced or interrupted according to a prespecified protocol if grade 2 or greater non-hematological toxicities or grade 3 or greater hematological toxicities occurred. Study treatment was discontinued if there was dose interruption of more than 21 days despite appropriate supportive care. Dose reduction was allowed up to two levels and the lowest dose was 25 mg in the continuous dosing regimen or 200 mg in the intermittent dosing regimen.
3. Evaluation
Safety profiles were evaluated by adverse events (AEs), vital sign, body weight, electrocardiogram, laboratory tests, ECOG performance status, and physical examinations. AEs were assessed and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), ver. 4.0.
Efficacy outcomes were measured using radiological assessments, including computed tomography/magnetic resonance imaging scans, and graded according to the Response Evaluation Criteria In Solid Tumors ver. 1.1. The same method of assessment and the same techniques were used to characterize each identified and reported lesion at the baseline and follow-up stages.
4. Dose-limiting toxicities
A DLT is defined as an AE with a causal relationship with the study treatment occurring in cycle 1 (Supplementary Methods). Unless they experienced a DLT, patients with drug compliance of < 75% during cycle 1 were excluded from the DLT evaluation and additional patients were enrolled.
5. PK analysis
Plasma was obtained for the determination of concentrations of OPB-111077 and its metabolites at days 1-4, 11, 18, and 24 in the continuous regimen cohort, and days 1, 8, 15, 16, 17, and 18 in the intermittent regimen cohort. Details of the pharmacokinetic analyses are summarized in the supplementary methods.
6. Statistical analysis
The primary endpoint was to determine the DLTs, MTD, RD, and safety profiles of OPB-111077 as a continuous or intermittent dose. The secondary endpoints include the PK parameters of OPB-111077, overall response rate, disease control rate (DCR) at 6 weeks and 12 weeks, progression-free survival (PFS), and overall survival (OS). The full analysis set included patients who received at least one dose of OPB-111077 and from whom post-treatment efficacy data were available. The safety analysis set consisted of all subjects that received at least one dose of OPB-111077.
7. Ethical statement
The study protocol was approved by the institutional review boards of participating centers, and all patients provided written informed consent before enrolment. The study was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice (ClinicalTrial. gov Identifier: NCT01942083).
Results
1. Patients
A total of 33 patients (19 for the continuous dosing cohort and 14 for the intermittent dosing cohort) were enrolled. Their baseline patient characteristics are summarized in Table 1. Cycle 1 of the study treatment was completed in 27 patients (81.8%) and DLTs were evaluable in 30 patients. All patients were Korean and had ECOG performance status of 0-1. Median age was 54 years (range, 23 to 69 years) and most patients (81.8%) were male. Hepatitis B virus infection was the most common etiology of HCC (n=26, 78.8%). Except for three patients (9.1%) with Child-Pugh B, all patients were classified as Child-Pugh A at baseline. All patients had extrahepatic metastasis and the most common metastatic sites were the lung (n=27, 82%) and lymph nodes (n=12, 36%). All patients previously received sorafenib and median 1 (range, 1 to 7) systemic chemotherapy regimen, including sorafenib, was administered prior to study enrollment.
Table 1.
Baseline patient characteristics
![]()
2. Dose limiting toxicities
One patient in the 250 mg continuous dosing cohort experienced a DLT of grade 3 dizziness. DLTs were not observed in the intermittent dosing cohort. The MTD was not identified in either the continuous or intermittent dosing cohorts. However, considering the PK profiles and increased frequencies of AEs with dose escalation, the RDs were determined to be 250 mg for continuous dosing and 600 mg for intermittent dosing.
3. Adverse events
All 33 patients were included in the safety analysis (Table 2). The duration of study treatment was median 22 days (range, 4 to 209 days); 40 days (range, 4 to 209 days) in the continuous dosing cohort and 12 days (range, 7 to 59) in the intermittent dosing cohort. There was no treatment-related death. All patients had at least one treatment-emergent AE (TEAE) and 5 patients (15.2%) had grade 3-4 TEAEs (10.5% in the continuous dosing cohort and 21.4% in the intermittent dosing cohort). The most frequently reported TEAEs were nausea (84.6%) and vomiting (48.5%). Although there was no dose-related trend in the frequency of TEAEs, there was an increase in the severity of nausea in the highest dose level (600 mg, 4-days on/3-days off), where most patients (6 of 7, 85.7%) experienced grade 2 nausea. The most frequent grade 3-4 TEAEs was thrombocytopenia (n=2, 6.1%). In the intermittent dosing cohort, vomiting (71.4% vs. 31.6%, p=0.02) and dizziness (64.3% vs. 26.3%, p=0.03) were significantly more common but decreased appetite was less common (14.3% vs. 52.6%) than in the continuous dosing cohort.
Table 2.
Adverse events occurring in > 10% of patients
![]()
TEAEs led to treatment discontinuation in three patients due to grade 3 abdominal pain and dizziness (both in the 200 mg continuous dosing cohort) and grade 4 thrombocytopenia (in the 300 mg intermittent dosing cohort). Dose reduction and interruption occurred in three (1 in the continuous and 2 in the intermittent dosing cohort) and five patients (1 in the continuous and 4 in the intermittent dosing cohort), respectively.
4. Efficacy
A total of 31 patients were included in the efficacy assessment. There were no patients who achieved complete or partial responses. Stable disease and progressive disease were the best responses in 13 (42%) and 18 (58%) patients, respectively. Five of 30 patients (16.7%) with measurable lesions showed reductions in the sizes of target lesions (Fig. 2). There was no significant difference in the rate of stable disease between dosing regimen (44.4% in the continuous dosing cohort vs. 38.5% in the intermittent dosing cohort; p > 0.99). The DCRs at 6 weeks and 12 weeks were 41.9% and 22.6%, respectively. There were also no dose-related trends in tumor response in either dosing regimen (S1 Table).
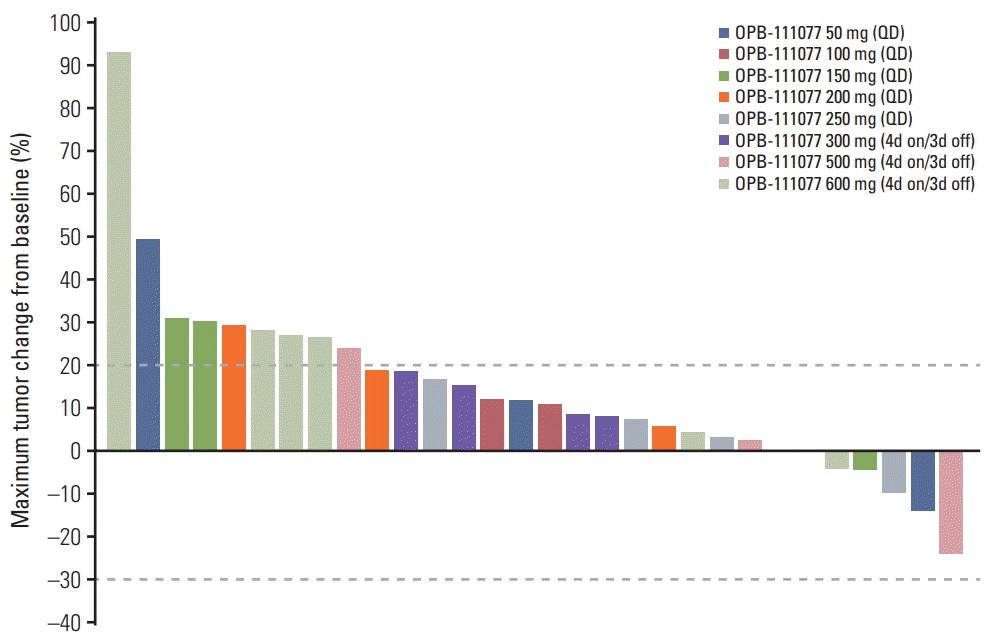
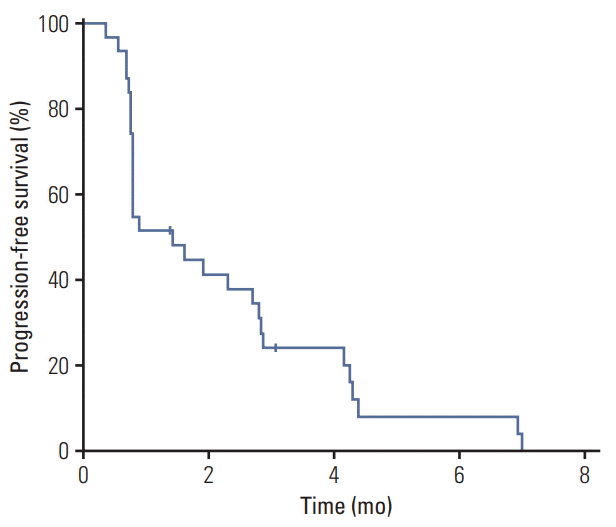
Median PFS was 1.4 months (95% confidence interval [CI], 0.8 to 2.8) and there was no significant difference between dosing regimens (median 1.6 months [95% CI, 0.8 to 4.2] in the continuous dosing cohort vs. 0.9 months [95% CI, 0.7 to 2.8] in the intermittent dosing cohort; p=0.21) (Fig. 3). Median OS was not reached at the time of analysis and 6-month and 1-year OS rates were 44.4% and 11.1%, respectively.
5. PK parameters
The results of PK analyses of the continuous dosing regimen and intermittent dosing regimen cohorts are listed in S2-S4 Tables. OPB-111077 was readily absorbed after both single and multiple doses, with median tmax values ranging from 1 to 4.96 hours across dose levels and dosing regimens. The half-life (t1/2), following multiple dose administration, ranged from 12.7 to 15.5 hours and was comparable across dosing levels and dosing regimens.
Discussion
This study was the phase 1 trial of the novel STAT3 inhibitor, OPB-111077, for patients with advanced HCC after failure of sorafenib. Two dosing schemes, continuous daily dosing and intermittent dosing (4-days on/3-days off administration), were tested to determine the DLTs, MTD, and RD. The safety profile, PK, and preliminary efficacy of each dosing schedule was also evaluated.
In this trial, the MTD was not determined in either the continuous or intermittent dosing cohorts. One patient experienced grade 3 dizziness, a prespecified DLT, in the 250 mg continuous dosing cohort, while no patients showed DLTs in the intermittent dosing cohorts. Considering the PK profiles and the increased frequency of AEs, particularly gastrointestinal symptoms, with dose escalation, the RDs of OPB-111077 for further evaluation in HCC populations were determined as 250 mg once daily for continuous dosing, and 600 mg once daily, 4-days on/3-days off, for intermittent dosing. These were consistent with the results of a previous phase I trial of OPB-111077 performed in United States, which included various types of cancers [20]. In this trial, the MTD of continuous daily OPB-111077 dosing was 250 mg once daily, and the observed DLTs were grade 3 nausea/vomiting at a dose of 400 mg once daily and grade 3 dizziness at a dose of 300 mg once daily.
PK analysis showed that OPB-111077 was readily absorbed after both single and multiple doses in both the continuous and intermittent dosing cohorts. Because of the small sample sizes, the data in this study were insufficient to reliably assess dose proportionality with inferential statistics. However, our results were in line with those of previous studies performed in United States [20].
OPB-111077 was well tolerated and showed favorable safety profiles. Most TEAEs were of grade 1 or 2 and manageable with appropriate supportive care. Nausea and vomiting were the most frequent TEAEs, and occurred in 84.6% and 48.5% of patients, respectively. Vomiting and dizziness were more commonly observed in the intermittent dosing cohorts, and decreased appetite in the continuous dosing cohorts. Grade 3-4 TEAEs occurred in 15.2% of patients and thrombocytopenia (6.1%), dizziness (3.0%), and fatigue (3.0%) were the most common severe toxicities. These safety profiles of OPB-111077 in HCC patients are consistent with the results of the previous phase 1 study of OPB-111077 in which continuous dosing for various cancer types showed that nausea (68.5%), vomiting (48%), fatigue (59.8%), and dizziness (31.5%) were common toxicities [20]. Previous trials that investigated other STAT3 inhibitors also reported that gastrointestinal symptoms, including nausea and vomiting, are the major TEAEs, with generally favorable safety profiles [17,18].
Despite 16.7% of patients with measurable lesions showing reduction in the sizes of target lesions, no patients achieved objective responses in this study, and the median PFS was only 1.4 months. There was no difference in efficacy between the different dosing schedules. Although based on a small number of patients, the current preliminary efficacy outcome did not justify the further evaluation of OPB-111077 in advanced HCC, considering that other investigational agents tested in randomized trials at the second-line setting after the failure of sorafenib showed median PFS (or time-to-progression) and OS of at least 3 months and 8 months, respectively [6,7,21-23]. Regorafenib, which has been approved as secondline therapy for sorafenib-progressed advanced HCC, showed a median PFS and OS of 3.1 months and 10.6 months, respectively [6]. An anti–PD-1 inhibitor, nivolumab, showed promising results in phase I/II trial with objective response rates of 15% and a median PFS of 4 months [7].
Despite the strong rationale for the investigation of STAT3 inhibition in HCC [9,11,14,24], the efficacy of small molecule STAT3 inhibitors, including ours, were limited [19]. In a previous Japanese study investigating another STAT3 inhibitor, OPB-31121, no patients showed objective responses and the median PFS was approximately 2 months in patients with HCC [19]. These findings indicate that STAT3 inhibitors, as a monotherapy, might not be effective in patients with HCC, and there are several hypotheses to explain these failures. First, there is the possibility that STAT3 inhibition was insufficient with these agents to produce anti-cancer activity. Because pharmacodynamic evaluation such as the measurement of STAT3 phosphorylation in peripheral blood was not done in these studies, it is not clear whether STAT3 was effectively targeted. Second, there is the lack of biomarkers for STAT3 inhibitors. Janus kinases (JAKs) are the upstream regulators of the JAK-STAT3 signaling pathway and ruxolitinib, a JAK2 inhibitor, is effective for patients with myelofibrosis and polycythemia vera in which JAK2 mutations are prevalent [25]. In contrast, there is no proven biomarker for STAT3 inhibitors and this limits the ability to stratify the patients who would benefit most from STAT3 inhibitors in the trial. Recent advances in understanding the role of STAT3 signaling in the development of HCC might help to revisit STAT3 inhibitors in subsets of HCC patients, such as those with FGF19-driven HCC or obesity-related HCC [9,12].
Although OPB-111077 showed limited efficacy as a monotherapy in patients with HCC, this agent might be further investigated as a combination therapy, considering its favorable safety profiles. Because the STAT3 signaling pathway has an immunomodulatory role in cancer development [26], the combination of OPB-111077 with anti–PD-1 inhibitors might be synergistic. The efficacy of OPB-111077 in combination with currently approved agents including sorafenib or regorafenib, or with promising investigational agents including FGFR inhibitors, might be also valuable to investigate.
In conclusion, OPB-111077 was well tolerated in patients with advanced HCC after failure of sorafenib, but only showed limited preliminary efficacy outcomes. Further investigation of the role of the STAT3 signaling pathway in HCC and the development of biomarkers for STAT3 inhibitors are warranted.
 XML Download
XML Download