

 PDF
PDF Citation
Citation Print
Print


Introduction
Kidney injury molecule-1 (KIM-1), also known as hepatitis A virus cellular receptor-1 and T-cell Ig mucin-1, is a transmembrane protein, which is not overexpressed in normal kidneys, but is significantly upregulated in damaged kidney tubular epithelial cells in toxic and ischemic acute kidney injury [1,2]. KIM-1 is comprised of an extracellular segment containing a mucin domain and six cysteine Ig-like domains and a cytosolic segment containing a tyrosine kinase phosphorylation motif [3]. The extracellular domain of KIM-1 is released from cells into the urine following kidney proximal tubular injury [3,4]. Of the different types of markers, KIM-1 has exhibited considerable superiority in the initial detection of acute kidney injury within 24 hours, which is earlier than the urine creatinine upsurge. These findings indicate that KIM-1 could serve as a potential tool for detection and treatment of acute kidney injury at early stages [5,6].
The frequency rates for all phases of renal cell carcinoma (RCC) have been rising consistently over the last three decades [7-9]. Partial initial warning signs result in late identification with metastases existing in nearly one third of patients at the time of diagnosis [10,11]. Thus, the availability of a specific and sensitive RCC biomarker and uncovering tumors prior to metastasis might significantly improve the prognosis of RCC. CD10, a cell surface metalloproteinase localized to the proximal nephron of the normal adult kidney [12], has been detected in nearly 90% of RCCs; however, it has also be identified in prostatic carcinomas, hepatocellular carcinomas, and urothelial carcinomas [13,14]. Notably, KIM-1 is overexpressed in most cases of RCC [15-17] and in other types of tumors including ovarian clear cell carcinoma [18] and lung cancer [19]. The extracellular domain of KIM-1 can be identified in the urine of patients with RCC and can thus be used as a biomarker for the detection of RCC [15-17].
Compared to large proteins or antibodies, small peptides possess the necessary properties to serve as imaging probes. They have competency of tissue diffusion because of their small sizes, fast clearance from blood circulation, giving less background signals, low levels of immunogenicity, and a lower manufacturing cost [20]. Additionally, it is relatively easy to chemically alter peptides to link them to imaging mate-rials or drugs. For example, octreotide, a somatostatin analogue radiolabeled with indium-111, is currently in clinical use for the detection of neuroendocrine tumors [21]. Screening of phage-displayed peptide libraries has effectively identified peptides that specifically bind to biomarkers at tumors and tumor vasculature. The three-amino-acid RGD peptide, a typical example of a phage display-identified peptide, targets tumor vascular endothelial cells through the αvβ3 integrin [22]. Using a phage displayed-peptide library, we previously identified a peptide that binds to the interleukin-4 receptor upregulated in cancer cells and utilized it for drug delivery to tumors [23,24]. In addition, we identified a peptide that binds to histone H1 exposed on the surface of apoptotic cells and used it for in vivo imaging of apoptosis following chemotherapy [25]. Furthermore, we identified a peptide that binds to myoglobin, a biomarker that is increased in the blood of patients with acute myocardial infarction [26]. In this study, we screened a phage displayed-peptide library to identify a peptide that binds to the KIM-1 protein and applied it in non-invasive in vivo imaging and detection of KIM-1–expressing tumors.
Materials and Methods
1. Cell culture
769-P human renal tumor cells and A549 human lung tumor cells were cultured in RMPI-1640 medium containing 10% fetal bovine serum (FBS). ACHN and A498 human renal tumor cells, HepG2 human liver tumor cells, and HEK-293 human embryonic kidney cells were grown in Dulbecco’s modified Eagle’s medium containing 10% FBS. All culture media were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin.
2. Biopanning of the phage-displayed peptide library for KIM-1–binding peptides
Enzyme-linked immunosorbent assay (ELISA) plates were coated with recombinant KIM-1 (Cloud-Clone Corp., Katy, TX) in 100 μL of a 10 μg/mL solution in 0.1 M NaHCO3 pH 9.6 at 4°C overnight or with bovine serum albumin (BSA; Bovogen, Melbourne, Australia) as a negative control. After washing the wells six times with Tris-buffered saline (TBS) containing 0.1% Tween (TBS-T), the plates were blocked with 1% of BSA in TBS at room temperature (RT) for 1 hour. Each biopanning round consisted of subtraction of phages nonspecifically bound to BSA and subsequent selection of phages bound to KIM-1, followed by amplification of the eluted phages. In the first round, an M13 phage library (New England Biolabs, Massachusetts, UK) at a titer of 1×1011 was added to BSA-coated wells and incubated at 4°C for 1 hour with gentle shaking for subtraction of phages that bound BSA. Following incubation, the unbound phages in the supernatant were collected and incubated with KIM-1–coated plates at 4°C for 1 hour with gentle shaking. Following incubation, the unbound phages were washed out using phosphate-buffered saline (PBS) containing 0.1% Tween (PBS-T). The concentration of Tween was gradually increased from 0.1% in the first round up to 0.5% in the fifth round, with a 0.1% increase in each round, in order to minimize the non-specific binding of phages. The KIM-1–bound phages were eluted by incubating the plates with 0.1 M glycine HCl (pH 3.1) at RT for 5 minutes and eluates were immediately neutralized with 1 M Tris-HCl (pH 9.1). Phage titration was performed by plaque assays. The phages were amplified using Escherichia coli ER2738 as a host and subjected to consecutive rounds of biopanning. The same amount of input was maintained in subsequent rounds.
3. DNA sequencing, amino acid sequence analysis, and peptide synthesis
Following five rounds of biopanning, phage clones were randomly picked and the inserted DNA was amplified by polymerase chain reaction for DNA sequencing. DNA samples of twenty individual clones were sequenced by Macrogen (Seoul, Korea). Peptide sequences corresponding to the DNA sequences were analyzed using the Clustal W program in order to align amino acid sequences and shared motifs and were also analyzed using the NCBI BLAST search to identify proteins with motifs homologous to the peptide sequences.
All peptides were synthesized by standard Fmoc method using a peptide synthesizer and purified by high-performance liquid chromatography to > 90% purity, and their mass were confirmed by matrix assisted laser desorption ionization-time of flight by Peptron Inc. (Daejeon, Korea). For fluorescence labeling, peptides were conjugated at the amino terminus with fluorescein isothiocyanate (FITC) dye, or with FPR675 near-infrared (NIR) fluorescence dye (BioActs, Incheon, Korea). The NSSSVDK peptide, a sequence present in T7 phage coat protein, was used as a control.
4. Immunofluorescence analysis for KIM-1 expression in cells
Cells (3×104 cells) were plated on the wells of 4-well chamber slides. After overnight culture, cells were fixed with 4% paraformaldehyde at RT for 10 minutes. Following washing, the cells were incubated with an anti–KIM-1 antibody (Novus Biologicals, Littleton, CO) at a 1:200 dilution in blocking buffer at RT for 2 hours and then with an Alexa594- or Alexa488-cojugated anti-rabbit secondary antibody (Thermo Fisher Scientific, Waltham, MA) at a 1:200 dilution in blocking buffer at RT for 1 hour. Cells were incubated with 4,6-diamidino-2- phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO) for nuclear staining. The chamber slides were then mounted with a ProLong anti-fade reagent (Thermo Fisher Scientific) and visualized with a fluorescent microscope (Zeiss, Oberkochen, Germany).
5. Phage protein- and cell-binding ELISA
For phage protein-binding ELISA, plates were coated with 100 μL of 10 μg/mL KIM-1 or BSA in 0.1 M NaHCO3 pH 9.6 at 4°C overnight. The plates were blocked with 1% BSA in TBS at RT for 1 hour. After washing the plates with TBS-T, the protein was incubated with 100 μL of each phage clone (1×108 plaque forming unit [pfu]/well) at RT for 1 hour. For phage cell-binding ELISA, cells (1×104 cells) were seeded in the wells of 96-well plates and allowed to grow overnight. Cells were blocked with culture medium containing 1% BSA at RT for 30 minutes. After washing the plates with PBS, cells were incubated with each phage clone (1×109 pfu/well) at 4°C for 1 hour, washed, and then incubated with the horseradish peroxidase-conjugated anti-M13 antibody (GE Healthhcare, Chicago, IL) diluted 1:3,000 in blocking buffer at 4°C for 1 hour. After washing the plates, 3,3',5,5'-tetramethylbenzidine substrate (Pierce, Rockland, IL) was added and incubated at RT for 10 minutes. The reaction was stopped using 2 M H2SO4 and plates were read at 450 nm using a microplate reader (Tecan, Zurich, Switzerland).
6. Flow cytometry analysis of peptide cellular binding
Cells were grown to 70% to 80% confluence, harvested, and suspended in a culture medium. Cells were blocked with culture medium containing 1% BSA at RT for 30 minutes and then incubated with 10 μM of a FITC-conjugated peptide in blocking buffer at 4°C for 1 hour. After washing, cells were subjected to flow cytometry (BD Bioscience, San Jose, CA).
7. Analysis of cellular binding of the NeutrAvidin/biotinpeptide multimer
To form the NeutrAvidin/biotin-peptide multimer, FITCconjugated NeutrAvidin (Thermo Fisher Scientific) at a concentration of 82.7 μM was incubated with biotin-conjugated peptide at a concentration of 330 μM at RT for 1 hour with rotation. The mixture was then dialyzed using a 15 kDa Tube-O-Dialyzer (G-biosciences, St. Louis, MO) in PBS at 4°C overnight. For immunofluorescence analysis, 3×104 cells were plated onto the wells of 4-well chamber slides. Following overnight incubation, cells were blocked with culture medium containing 1% BSA at RT for 30 minutes and then incubated with 10 μM of the NeutrAvidin/biotin-peptide multimer in blocking buffer at 4°C for 1 hour. Cells were fixed with 4% paraformaldehyde at RT for 10 minutes. For co-localization, cells were incubated with the anti–KIM-1 antibody at a 1:200 dilution in blocking buffer at RT for 2 hours and then incubated with an Alexa594-conjugated antirabbit secondary antibody at a 1:200 dilution in blocking buffer at RT for 1 hour. Cells were incubated with DAPI for nuclear staining and with an anti-fade reagent prior to observation using a fluorescence microscope (Zeiss). For flow cytometry analysis, cells were harvested, blocked, and then incubated with 10 μM of the NeutrAvidin/biotin-peptide multimer in blocking buffer at 4°C for 1 hour. For competition of binding, different concentrations of the anti–KIM-1 antibody were pre-incubated at RT for 2 hours prior to incubation with the NeutrAvidin/biotin-peptide multimer. Following incubation, cells were subjected to flow cytometry (BD Bioscience).
8. Fluorometric analysis of peptide and NeutrAvidin/biotin-peptide multimer binding to KIM-1
Black colored microplate wells (SPL Life Science, Pocheon, Korea) were coated with 100 μL of 10 μg/mL BSA or KIM-1 in 0.1 M NaHCO3, pH 9.6 and incubated at 4°C, overnight. The plates were blocked at RT for 1 hour with a blocking buffer (1% BSA in PBS) and incubated with 100 μL of 5 μM FITC-NeutrAvidin/biotin-peptide multimer in blocking buffer at RT for 1 hour. After washing the plates, fluorescence intensity was measured using a fluorometer.
9. Serum stability assays
Mouse blood was collected and allowed to clot. Serum was obtained by centrifuging the blood clots twice at 4°C followed by filtration through a 0.22 μm filter. Each peptide (100 μg in 50 μL of PBS) was incubated with 50 μL of filtered serum at 37°C for the indicated time periods. The incubated samples were diluted 100-fold and fractionated by C18 reverse phase fast performance liquid chromatography with a linear gradient of acetonitrile. The peptide peak was collected and analyzed.
10. Cytotoxicity assays
Cells (1×104 cells) were seeded in the wells of 96-well plates and allowed to grow overnight. Cells were incubated with the indicated concentration of peptide at 37°C for 24 hours. Cells were then incubated with 10 μL of the CCK-8 dye (Dojindo, Kumamoto, Japan) mixed with 100 μL of culture medium. Absorbance was measured with a microplate reader at 450 nm.
11. In vivo imaging and histological analysis of tumor homing by peptide
Tumor cells (5×106 cells) were subcutaneously injected into the right flank of 6-week-old BALB/c nu/nu female mice. Tumor-bearing mice were injected via the tail vein with 20 nmol of flamma675 NIR dye (BioActs, Incheon, Korea)-conjugated peptide. In vivo fluorescence images were obtained using an IVIS imaging system (Perkin Elmer, Waltham, MA) at 2, 4, 8, and 24 hours after injection. At the end of in vivo imaging, tumor and other organs were isolated and ex vivo fluorescence images were taken. After taking the ex vivo images, the tumors and organs were fixed with 4% paraformaldehyde for 2 hours and frozen for cryosection. Tissue slides were incubated with the anti–KIM-1 antibody (1:200 dilution) at RT for 2 hours and with the Alexa594 anti-rabbit secondary antibody (1:200 dilution) at RT for 1 hour. Tissues were mounted using mounting medium containing DAPI for nuclear staining and observed with a fluorescence microscope.
Results
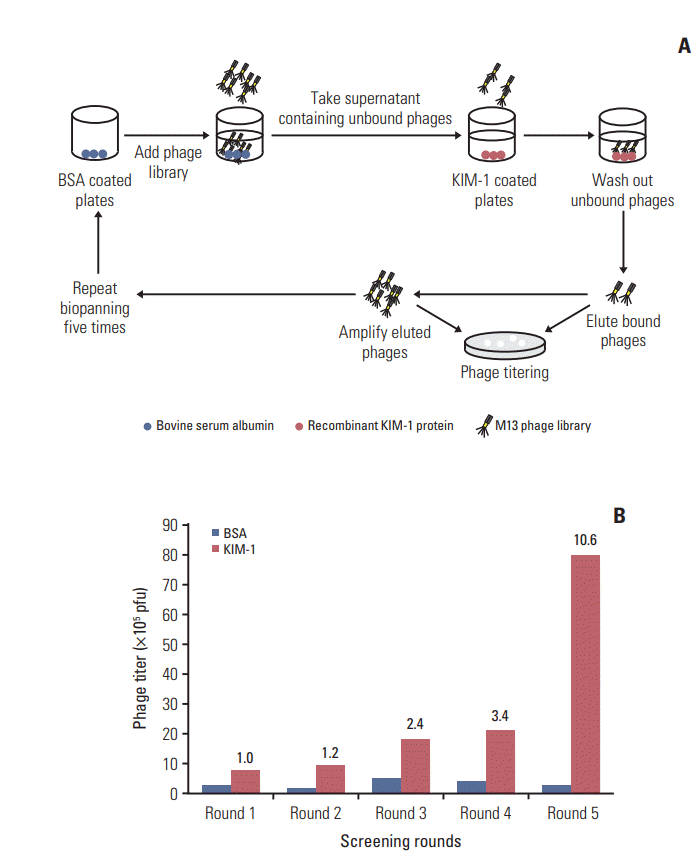
1. Screening of a phage-displayed peptide library for peptides that bind to the KIM-1 protein
An M13 phage library displaying CX7C random peptides was screened in vitro to identify phage clones that bind to KIM-1 protein-coated plates. The overall screening scheme is shown in Fig. 1A. Following five rounds of screening, the phage titer of the fifth round was enriched approximately 10-fold compared to that of the first round (Fig. 1B). Twenty phage clones were randomly picked from the third, fourth, and fifth rounds and the peptide-coding DNA inserts of the phage clones were sequenced (Table 1). Four phage clones displaying the CNWMINKEC, CVPSKPGLC, CPKGDENTC, and CPTSQRDNC sequences were chosen for further evaluation, as they occurred more frequently than other phage clones.
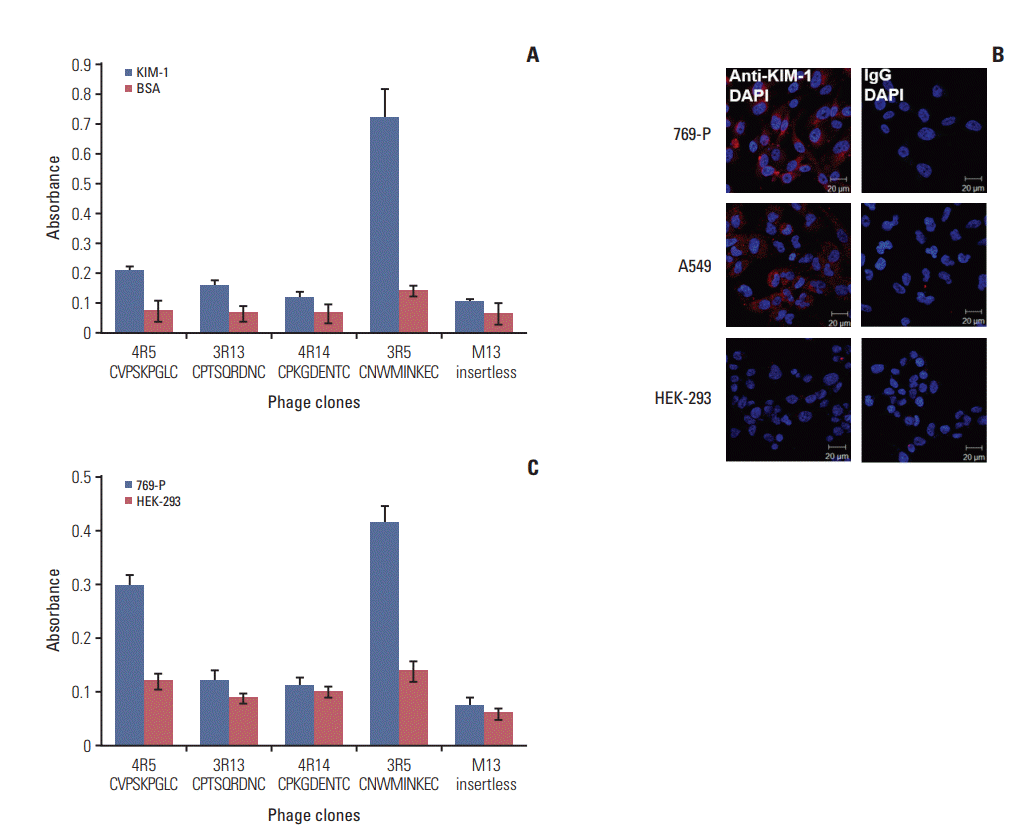
2. Binding of phage clones to the KIM-1 protein and KIM-1–overexpressing cells
The binding activity of individual phage clones to the KIM-1 protein was examined by phage-binding ELISA. Compared to the other phage clones, the CNWMINKEC peptide phage clone showed higher levels of binding to the KIM-1 protein relative to BSA (Fig. 2A). Next, we examined the binding of the phage clones to KIM-1–overexpressing cells. Immunofluorescence analysis showed that 769-P renal cancer cells and A549 lung cancer cells expressed higher levels of KIM-1 on the cell surface than HEK-293 normal cells (Fig. 2B). The CNWMINKEC-phage clone and CVPSKPGLC-phage clone demonstrated higher levels of binding to KIM-1–overexpressing 769-P cells than to KIM-1–low expressing HEK-293 cells compared to the other phage clones and the control M13 insertless phage clone (Fig. 2C).
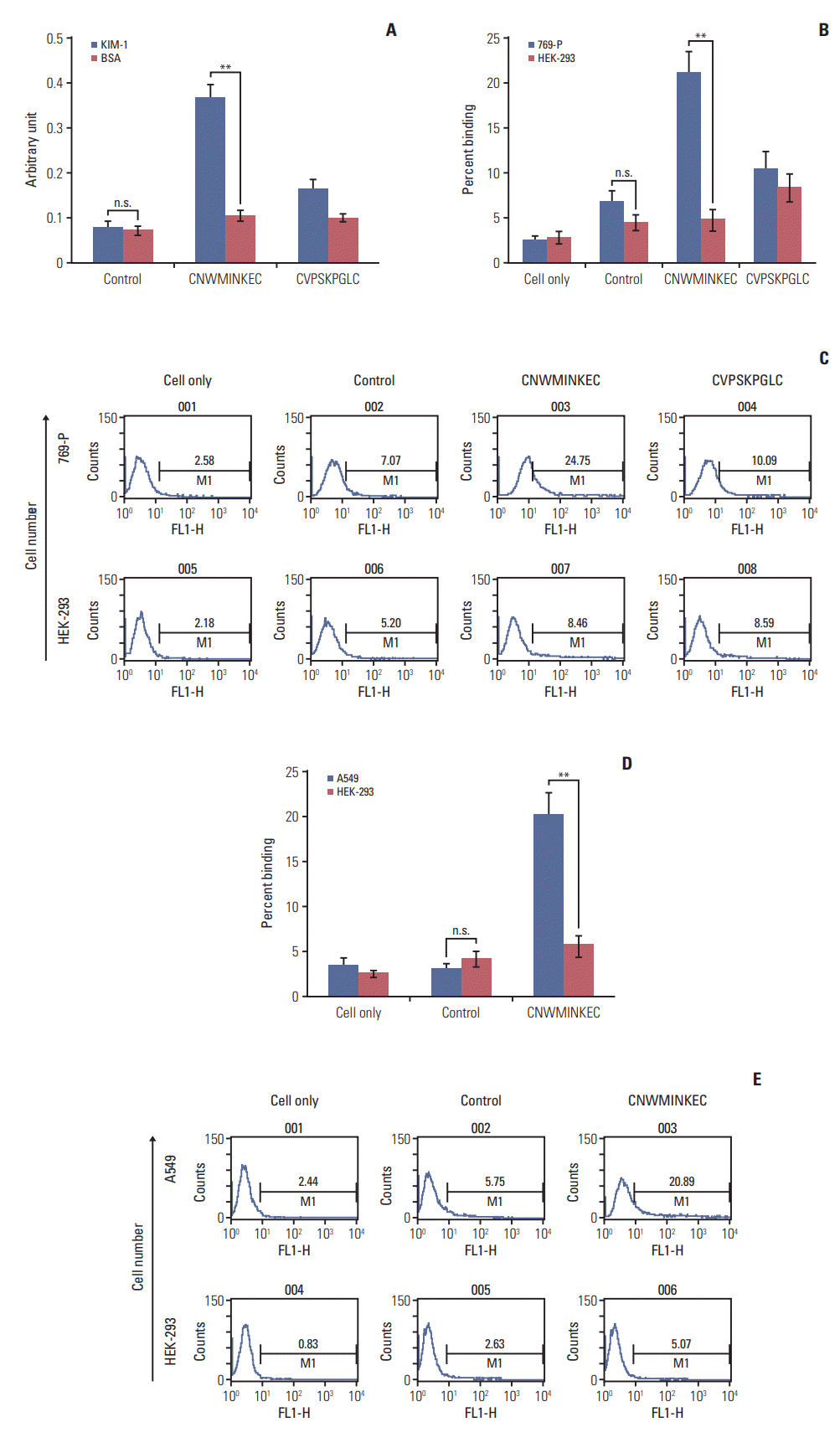
3. Binding of peptides to the KIM-1 protein and KIM-1–expressing cells
To examine the binding of the CNWMINKEC and CVPSKPGLC peptides to the KIM-1 protein and KIM-1–overexpressing cells, the peptides were synthesized and conjugated with a FITC green fluorescent dye at the amino terminus. Analysis of peptide binding using a fluorometer showed that the CNWMINKEC peptide bound to the KIM-1 protein at higher levels than to BSA (Fig. 3A). The binding of the CVPSKPGLC and control peptides to the KIM-1 protein were similar to their binding to BSA and were much lower than the CNWMINKEC peptide (Fig. 3A). Flow cytometry analysis demonstrated that the percent binding of the CNWMINKEC peptide to 769-P cells was approximately four-fold higher than to HEK-293 cells (21% and 5%, respectively) (Fig. 3B), while the percent binding of CVPSKPGLC to 769-P cells was similar to the binding of HEK-293 cells (10% and 8%, respectively) (Fig. 3B and C). In addition, the percent binding of CNWMINKEC to A549 lung tumor cells was approximately three-fold higher than to HEK-293 cells (approximately 20% and 6%, respectively) (Fig. 3D and E). These results indicate that the CNWMINKEC peptide preferentially binds to the KIM-1 protein and KIM-1–overexpressing cells compared to albumin and KIM-1–low expressing cells, respectively.
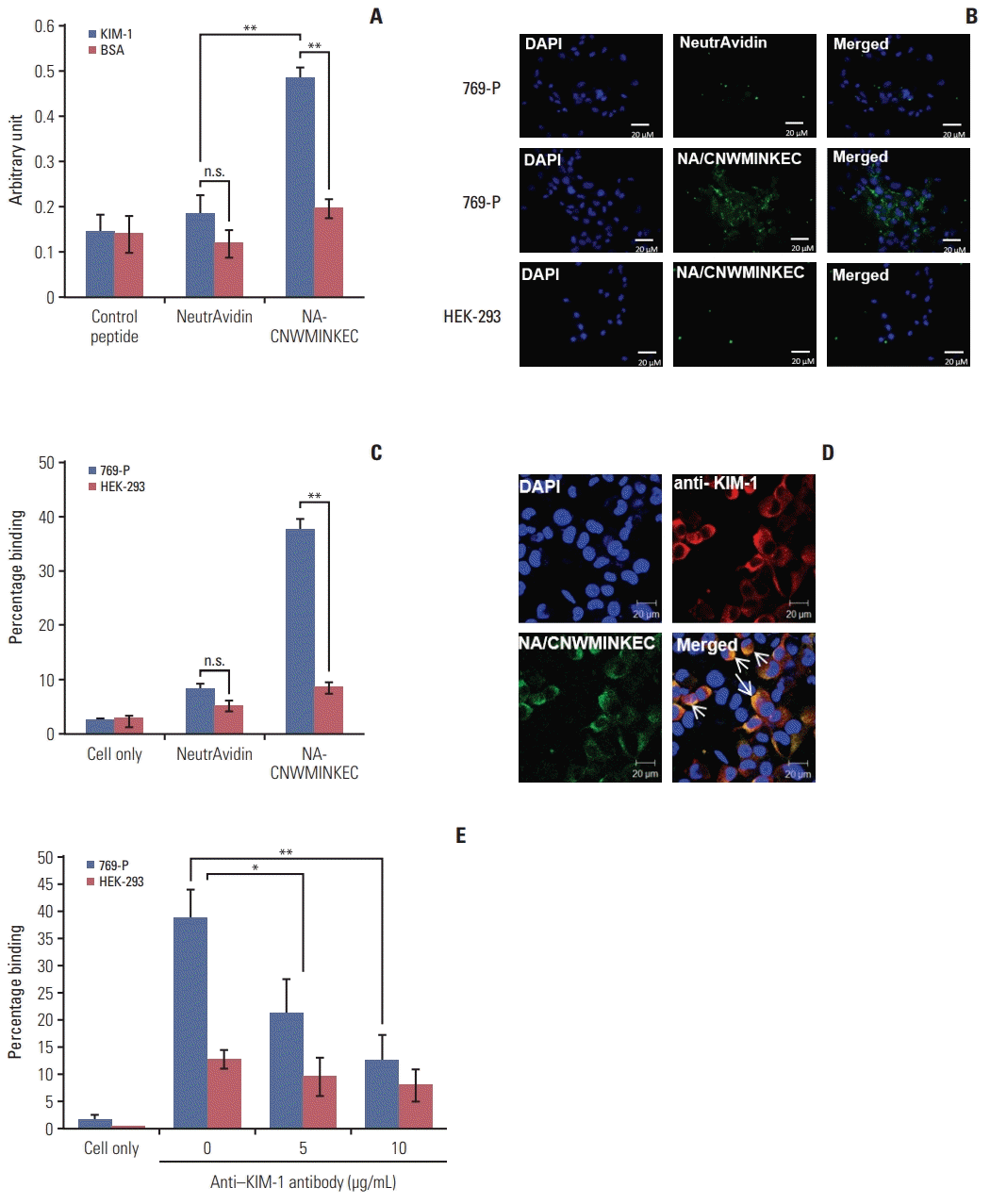
4. Binding of the CNWMINKEC peptide multimer to the KIM-1 protein and KIM-1–overexpressing cells
Biotin-labeled CNWMINKEC peptide was incubated with NeutrAvidin to form a multimeric complex using the biotinavidin interaction. The binding of the NeutrAvidin/biotin-CNWMINKEC multimer to KIM-1 protein was higher than to the BSA control, while NeutrAvidin itself showed only minimal levels of binding to both proteins (Fig. 4A). Immunofluorescence analysis demonstrated that the binding of the NeutrAvidin/biotin-CNWMINKEC multimer to 769-P cells was higher than to HEK-293 cells, while the binding of NeutrAvidin to 769-P cells was negligible (Fig. 4B). The percent binding of the NeutrAvidin/biotin-CNWMINKEC multimer to 769-P cells was approximately four-fold higher than to HEK-293 cells, as determined by flow cytometry (38% and 9%, respectively) (Fig. 4C). The NeutrAvidin/biotin-CNWMINKEC multimer co-localized with KIM-1 on 769-P cells, as observed by confocal fluorescence microscopy following co-staining with the anti–KIM-1 antibody (Fig. 4D). Furthermore, pre-treatment of 769-P cells with the anti–KIM-1 antibody reduced the subsequent binding of the NeutrAvidin/biotin-CNWMINKEC multimer in a dose-dependent manner, while minimal effect was observed in HEK-293 cells (Fig. 4E). These results suggest that the binding of CNWMINKEC to 769-P cells is specifically mediated by KIM-1.
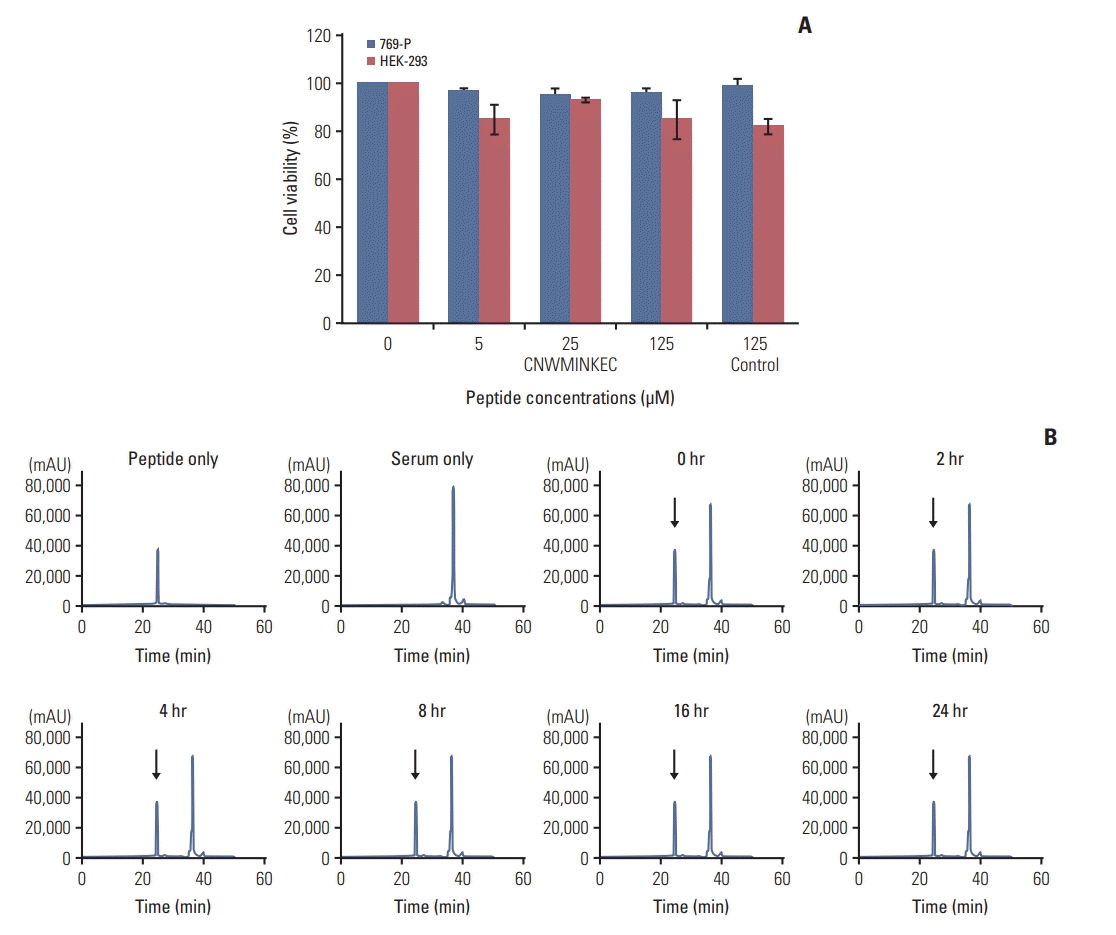
5. Cytotoxicity and serum stability of the CNWMINKEC peptide
To examine the non-specific cytotoxicity of the CNWMINKEC peptide, 769-P and HEK-293 cells were incubated with different concentrations of the CNWMINKEC peptide for 24 hours. Treatment of the cells with the CNWMINKEC peptide did not significantly affect cell viability (Fig. 5A), suggesting that the CNWMINKEC peptide itself has no cytotoxicity. To examine the serum stability of the CNWMINKEC peptide, it was incubated with mouse serum for up to 24 hours and the amount of the peptide present over a period of time was analyzed. The peptide peak was separable from the non-specific serum peaks and was not significantly reduced up to 24 hours (Fig. 5B). These results suggest that the CNWMINKEC peptide is stable in the serum for up to 24 hours.
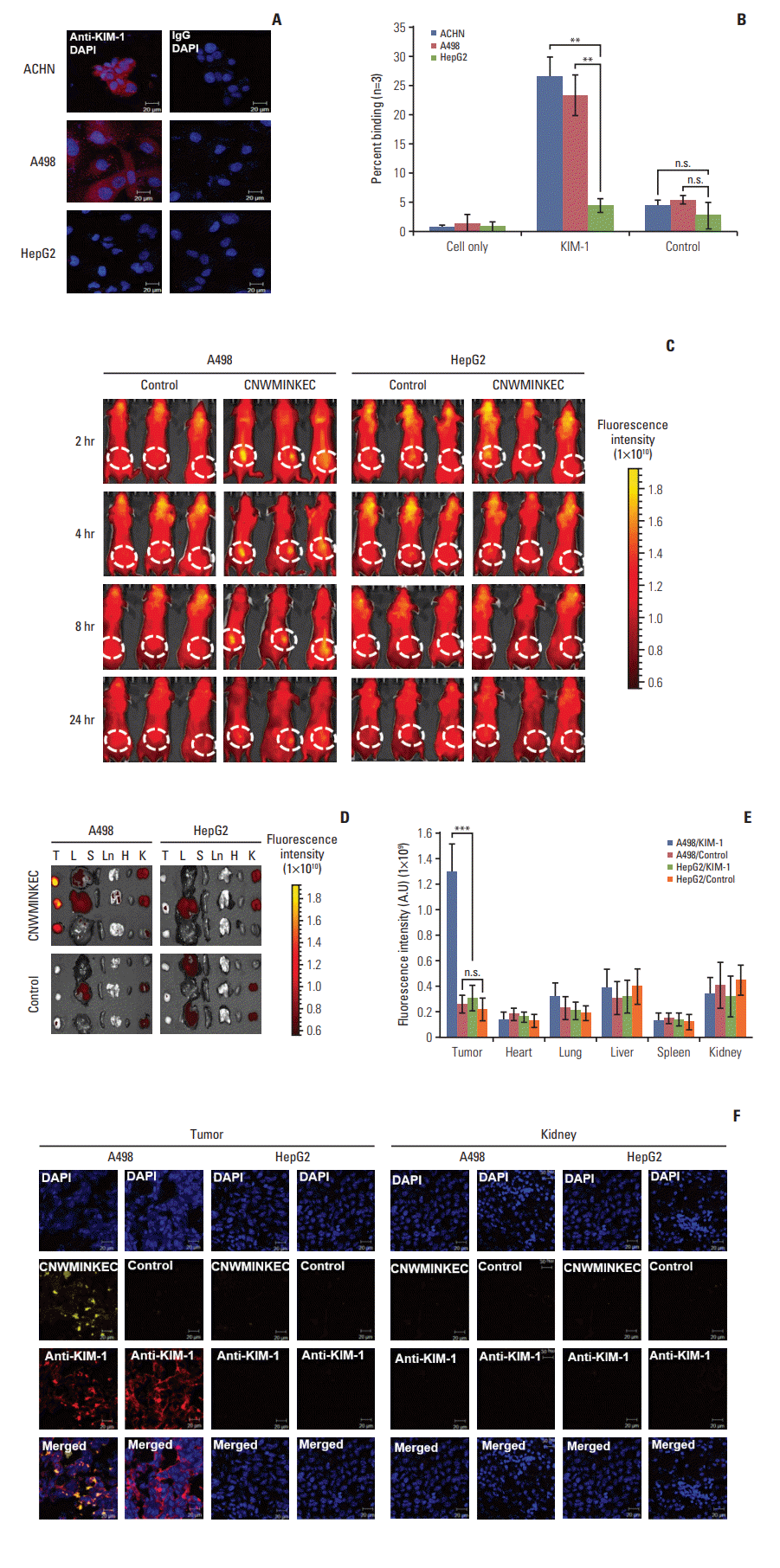
6. In vivo imaging and detection of KIM-1–overexpressing tumor using the CNWMINKEC peptide
As 769-P cells do not form tumors in mice, we used A498 cells, another renal tumor cell line, as a KIM-1–overexpressing tumor model. In addition, we used HepG2 liver tumor cells as a KIM-1–low expressing tumor model. Immunofluorescence analysis showed that A498 and ACHN renal tumor cells highly express KIM-1, while HepG2 cells little express KIM-1 (Fig. 6A). The percent binding of the CNWMINKEC peptide to A498 and ACHN cells was approximately six-fold higher than that to HepG2 cells (23% and 26% vs. 4%) (Fig. 6B). Mice bearing A498 or HepG2 tumor were intravenously injected with the flamma675 NIR fluorescence dye-labelled CNWMINKEC and control peptide. Whole body fluorescence imaging showed enhanced fluorescence signals at KIM-1–overexpressing A498 tumor by the homing and accumulation of the CNWMINKEC peptide at 2-, 4-, and 8-hour post injection with a decline at 24 hours, while minimal signals were detected at A498 tumor by the control peptide (Fig. 6C). Only minimal or background signals were detected with either the CNWMINKEC or control peptide at KIM-1–low expressing HepG2 tumor (Fig. 6C). Ex vivo fluorescence images of isolated organs further demonstrated higher levels of fluorescence signals at A498 tumor in mice injected with the CNWMINKEC peptide compared to control peptide, whereas little signals were detected at HepG2 tumor (Fig. 6D and E). Compared to A498 tumor, much lower levels of fluorescence signals were observed at the liver, lungs, and kidneys of mice injected with either the CNWMINKEC or control peptide (Fig. 6D and E), suggesting selective homing of the CNWMINKEC peptide to KIM-1–overexpressing tumor. In addition, immunohistochemical examination of tissues demonstrated that the CNWMINKEC peptide co-localized with KIM-1 at tumor tissues, as detected by staining with an anti–KIM-1 antibody (Fig. 6F). Accumulation of either the CNWMINKEC or control peptide in kidney tissues was not detected (Fig. 6F), suggesting that the ex vivo fluorescence signals at the kidney were not due to accumulation in the kidney, but rather to urinary excretion of the peptides.
Discussion
By screening a phage displayed-peptide library, we identified the CNWMINKEC peptide that was able to bind to the KIM-1 protein and KIM-1–overexpressing cells. The CNWMINKEC peptide selectively bound to purified KIM-1 protein over albumin. In addition, it bound to KIM-1–overexpressing 769-P, A498, and ACHN renal tumor cells as well as A549 lung tumor cells at higher levels than to HEK-293 normal cells and KIM-1–low expressing HepG2 liver tumor cells. The biotin-CNWMINKEC peptide multimer, generated via the interaction between biotin and avidin, also selectively bound to the KIM-1 protein and KIM-1–overxpressing tumor cells. The CNWMINKEC peptide multimer co-localized with KIM-1 on KIM-1–expressing cells, and this binding was inhibited by pre-treating the cells with an anti–KIM-1 antibody. These results indicate that the binding of the CNWMINKEC peptide to the KIM-1–overexpressing cells is mediated by KIM-1. The CNWMINKEC peptide was accumulated in KIM-1–overexpressing, but not in KIM-1–low expressing, tumor tissues in vivo, while only minimal accumulation was observed in other organs including the liver and kidneys. The CNWMINKEC peptide did not cause non-specific cytotoxicity in cells and was stable for up to 24 hours in serum. These findings suggest that the CNWMINKEC peptide is a promising probe for non-invasive in vivo imaging and detection of KIM-1–overexpressing tumors.
KIM-1 has been used as an immunohistochemical marker for the diagnosis of certain types of tumors, including RCC and ovarian clear cell carcinoma, in which KIM-1 expression is up-regulated [15,18,19]. Moreover, KIM-1 expression is related to a more malignant phenotype of RCC and shedding of the ectodomain has been associated with tumor advancement [27]. Yet, this does not provide information regarding the location of primary and metastatic RCC. Positron emission tomography (PET) that is based on the uptake of 18F-fluoro-2-deoxy-2-D-glucose (FDG) by tumor cells has been extensively used for the detection of tumors. Unlike to most other tumors, however, FDG-based PET is limited for primary RCC due to physiological excretion of FDG through the urine and reabsorption in the kidney tubule, which causes non-specific signals and decreases contrast between tumor lesions and normal tissues of the kidney [28]. In this study, we found that the CNWMINKEC peptide was almost not detected in normal tissues of the kidney in mice when examined 24 hours after the injection. These results suggest that the KIM-1–binding CNWMINKEC peptide holds a potential as an imaging probe for the detection of primary as well as metastatic RCC.
In addition to its use as a diagnostic biomarker, KIM-1 is a useful therapeutic target. An anti–KIM-1 antibody-drug conjugate has been developed as potential therapy for ovarian, lung, and renal cell carcinomas expressing KIM-1 [19,29]. Owing to the efficient tissue penetration ability of smallsized peptides, the CNWMINKEC peptide could be used as a promising targeting moiety for a peptide-drug conjugate for cancer therapy. Moreover, drug-loaded nanoparticles labeled with the CNWMINKEC peptide multimer may be delivered to KIM-1–overexpressing tumors with enhanced specificity as well as possess enhanced therapeutic efficacy.
A protein database search revealed several proteins homologous to the CNWMINKEC peptide sequence. For example, CNWMINKEC has homology with the 959WMIDSEC965 sequence of the receptor tyrosine kinase ErbB-2 isoform, the 167NWLINEC173 sequence of the transmembrane protease serine 11D, and the 2192MINKE2196 sequence of the WD repeatcontaining protein 87 isoform 1. Further studies on the interaction of KIM-1 with the proteins mentioned above may provide useful information for identifying a potential endogenous ligand that binds to KIM-1 up-regulated in tumor and kidney injury.
To the best of our knowledge, this is the first report to identify a phage library-derived peptide that binds to KIM-1 protein and use it for non-invasive detection of KIM-1–overexpressing tumors in vivo. In addition to its use as an imaging probe, the peptide could be conjugated with nanoparticles containing therapeutic agents, or directly conjugated with a drug (peptide-drug conjugate) and used for selective drug delivery to tumor tissues, thus enhancing the local concentration of the drug(s), while reducing their side effects. In this context, the KIM-1–binding CNWMINKEC peptide could be a cost-effective alternative to anti–KIM-1 antibodies as a diagnostic and therapeutic tool for the detection and treatment of cancers overexpressing KIM-1.
 XML Download
XML Download