

 PDF
PDF Citation
Citation Print
Print


Introduction
East Asian never-smokers have a higher prevalence of epidermal growth factor receptor (EGFR) mutations compared to their North American/European counterparts [1]. EGFR–tyrosine kinase inhibitors (TKIs) are effective clinical therapies for EGFR-mutant non-small cell lung cancer (NSCLC) patients [2]. Osimertinib, a third-generation EGFR TKI, has proven effective against T790M mutation. However, the vast majority of patients acquire resistance following successful treatment.
Many studies have reported that the EGFR C797S mutation is the most common mechanism of resistance to third-generation EGFR TKIs, including osimertinib (AZD9291) [3,4]. Here we report two Korean cases of a rare mutation—EGFR C797G, L718Q—as a mechanism of acquired resistance to osimertinib.
The medical records of these patients were retrospectively reviewed with the approval of the Institutional Review Board of the Samsung Medical center (IRB No. SMC 2018-05-076) with the waiver for informed consent.
Case Report
1. Case 1
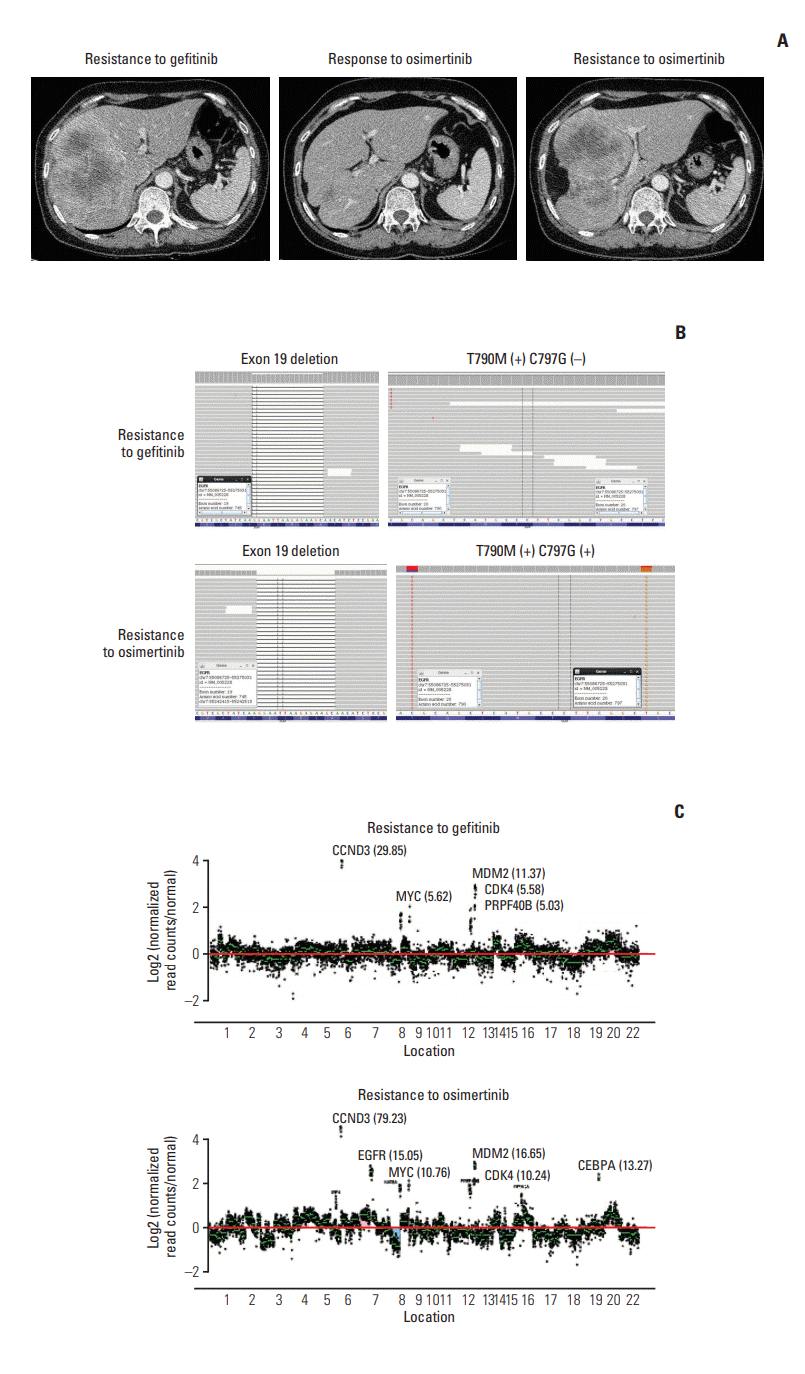
A 59-year-old female never-smoker was diagnosed with EGFR exon 19 deletion-positive metastatic lung adenocarcinoma, which was poorly differentiated with squamoid differentiation, in March 2015. The pathology result was initially reported as squamous cell carcinoma, and the patient underwent chemotherapy with four cycles of gemcitabine and carboplatin, leading to partial remission of the primary lesion in the left lower lobe. After seven months off treatment, the primary tumor in the left lower lobe, pleural seeding, and hepatic metastasis had increased in extent. The patient received gefitinib from January 2016 to December 2016, until progression of hepatic metastasis was demonstrated. Both real-time polymerase chain reaction (PCR) and CancerSCAN [5] of the post-gefinitib liver biopsy revealed an EGFR T790M mutation.
After 8 weeks of osimertinib treatment, scans demonstrated a partial response; however, the patient developed hepatic metastasis progression after 43 weeks of osimertinib (Fig. 1A). CancerSCAN [5] of the liver biopsy at the time of progression revealed an EGFR C797G mutation at an allele frequency of 72%, including the known T790M mutation (73%) in cis and an exon 19 deletion (87%), as well as focal amplification of EGFR (15.0) (Fig. 1B and C). As opposed to other preexisting N-ethyl-N-nitrosourea (ENU) mutagenesis studies, there was no coexisting EGFR C797S mutation. EGFR C797G mutation was the sole mutation with high minor allele frequency.
2. Case 2
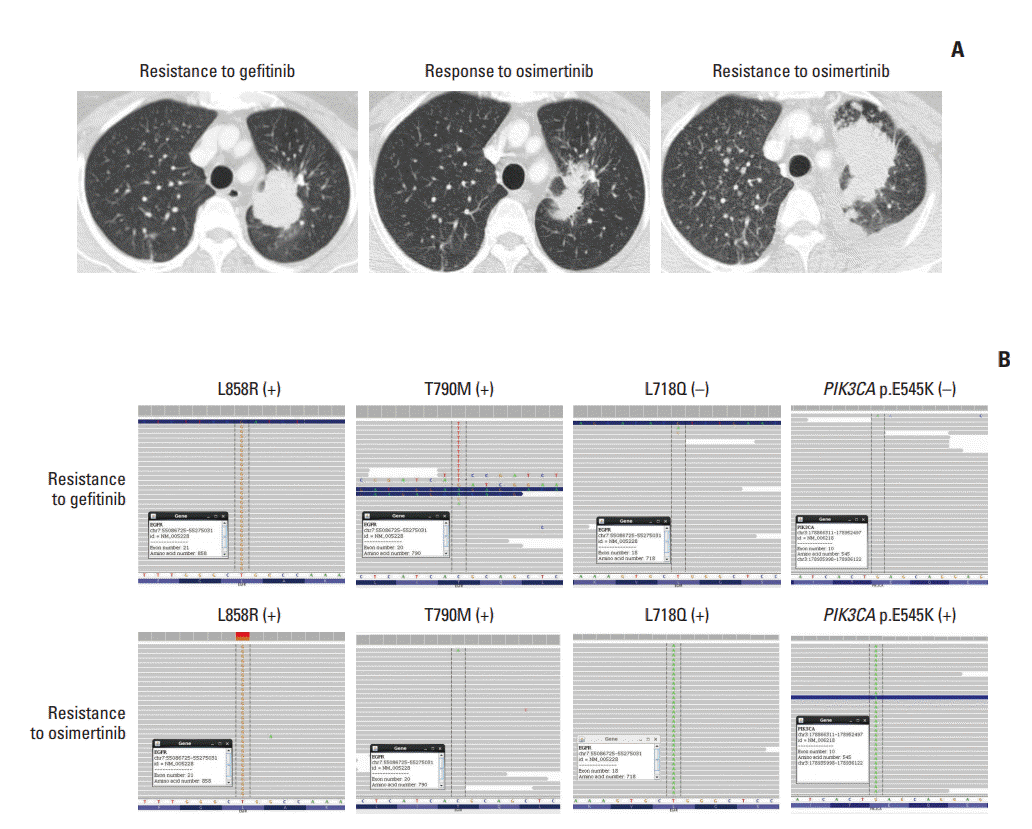
A 53-year-old female never-smoker was diagnosed with advanced lung adenocarcinoma in October 2013. A pleural tumor specimen showed a missense mutation on exon 21, and the patient received gefitinib for 33 months as the first-line therapy. While continuing gefitinib, the patient underwent whole-brain irradiation for multiple brain metastases and leptomeningeal seeding. Progressive disease was detected, and a new pulmonary biopsy identified T790M mutation. The patient received 10 months of osimertinib from August 2016 to May 2017, until disease progression was confirmed (Fig. 2A).
At the time of systemic progression, cancerSCAN [5] was performed on the lung biopsy specimen and identified an EGFR L718Q mutation at an allele frequency of 7% and a preexisting EGFR L858R at an allele frequency of 38%, but loss of the T790M mutation (Fig. 2B). Concomitant PIK3CA E545K at an allele frequency of 12.9% was also identified.
The patient received afatinib, the most effective EGFR inhibitor in cell line models harboring L858R along with L718Q mutation [4], which induced partial remission after 4 weeks of therapy; however, she demonstrated systemic progression after 12 weeks of therapy.
Discussion
Herein, we describe two Korean patients with a rare EGFR mutation after cancer treatment with osimertinib, a thirdgeneration EGFR TKI.
The EGFR C797S mutation is detected in ~40% of EGFR mutant NSCLC patients with T790M who develop acquired resistance to osimertinib [4]. However, emerging clinical data have revealed a high degree of clonal heterogeneity in the resistance mechanism, including non-EGFR–driven mutations.
Pre- and post-osimertinib genomic profiling results of cases 1 and 2 is presented in S1 and S2 Tables.
In case 1, although the variant allele frequency of EGFR T790M was detected pre-osimertinib CancerSCAN [5] at a low rate (0.4%) (S1 Table), it was simultaneously detected on real-time PCR. In refractory tumors, a driver mutation that arises in response to treatment may be present with a low variant allele frequency [5]. Considering the durable response to osimertinib, EGFR T790M appears to be the most reasonable mechanism of resistance to gefitinib.
In case 2, pathway alterations beyond EGFRwere reported from post-osimertinib CancerSCAN of the lung specimen, most notably PIK3CA E545K (12.90%) (S2 Table).
Co-occurrence of the PIK3CAmutation with EGFR aberrations are frequently reported [6], and an in vitro study has shown that PIK3CA oncogenes are sufficient to promote continued phosphoinositide 3-kinase (PI3K) signaling in EGFRmutant cancer cells treated with EGFR TKIs [7]. Thus, cancer with PIK3CA oncogenic mutations exhibit attenuated responses to EGFR inhibitors, suggesting that these mutations the mechanism of acquired resistance [8]. Such acquired resistance possibly resulted in the short response duration observed with afatinib in case 2.
Although EGFR mutation still remains the principal “mutational driver,” there is increasing evidence of complexity in the resistance mechanism to third-generation EGFR TKIs. Dactolisib, a dual PI3K/mammalian target of rapamycin (mTOR) inhibitor, has proven effective in inhibiting gefintib-resistant tumor growth by downregulating PI3K/anaplastic lymphoma kinase (ALK)/mTOR phosphorylation [9]. However, a phase II trial of MK-2206 (AKT inhibitor for PI3K/ALK/mTOR pathway) and erlotinib in NSCLC patients who progressed on erlotinib, showed a frustrating Response Evaluation Criteria in Solid Tumor response rate 9% at 12 weeks in the EGFR-mutant group.
Our cases highlight the importance of a genomic profiling approach to detect alterations associated with uncommon mechanisms of acquired resistance to targeted therapies. Further studies are warranted on novel or combination agents to overcome the rare acquired resistances.
 XML Download
XML Download