

 PDF
PDF Citation
Citation Print
Print


Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide and is responsible for a quarter of a million deaths annually [1].
Stimulation of cells for proliferation, differentiation, and apoptosis needs extracellular signals, which are relayed to the nucleus where they alter the pattern of gene expression. Mitogen-activated protein kinases (MAPK) are important components of intracellular signaling pathways that, when activated, relay signals further in response to many extracellular stimuli such as growth factors, osmotic stress, etc., thus leading to inflammatory responses and eventually into development of cancer.
p38 MAPK is a family of four isoforms: p38α, p38β, p38γ, and p38δ. All the isoforms are homologous and are activated through the dual phosphorylation of tyrosine (Tyr) and threonine (Thr) residues in conserved Thr-X-Tyr motif mediated by the MAPK. Moreover, the p38 MAPK family is divided into two subgroups based on sequence similarity, cellular expression patterns, and substrate specificity. p38α and β represent one subgroup with sequence identity of 75%, whereas p38γ and δ represent another subgroup with sequence identity of 70% approximately. p38α and β are the most ubiquitously expressed in all cells. In contrast, the sequences of p38γ and δ are different and have 62% and 61% sequence homology to p38α, respectively: this subgroup is expressed in specific tissues only [2]. The p38 MAPK pathway is involved in the process of inflammation, cell differentiation, cell growth, and apoptosis. It is directly involved in the production of pro-inflammatory cytokines such as tumor necrosis factor α and interleukin 1β. Overproduction of these cytokines has been implicated in a variety of diseases that have an inflammatory component. The specific role of p38 MAPK-regulated inflammatory response is to facilitate tumor growth.
p38δ has recently emerged as a potential disease-specific drug target, as it is reported to be critically involved in numerous inflammatory diseases and cancers. Several p38α inhibitors have been identified, and pyridinyle imidazole inhibitor SB203580 is found to be a very specific p38α inhibitor. This study aims to evaluate the expression level of cell signaling molecules in HNSCC cases and to develop a peptide inhibitor that targets the significant molecule for such cases.
Materials and Methods
1. Patient selection and treatment
Sixty HNSCC cases and 52 ethically matched healthy controls were recruited in this study. The blood samples were collected from Head and Neck Cancer Clinic, Dr. B.R.A. Institute Rotary Cancer Hospital (IRCH), All India Institute of Medical Sciences (AIIMS), New Delhi, India after obtaining their written informed consent. The diagnosis of the disease was based on the staging criteria given by the American Joint Committee on Cancer. All cases were carefully examined by clinicians of AIIMS, and their medical history was determined through a questionnaire. They also underwent complete physical examination (extent, stage, and possible treatment options). Combined modality treatment (CMT) was chosen for advanced HNSCC cases. CMT is a treatment that encompasses surgery followed by post-operative radiotherapy or concurrent chemoradiotherapy on the basis of patient’s age, disease stage, and performance status. Cisplatin is commonly used for chemotherapy, with scheduled dose of 40 mg/m2 that is controlled for five cycles, with one cycle per week. Radiotherapy schedule usually consists of 2 Gy per fraction and is given for 5 days in a week, for a total duration of 6 to 7 weeks on Co60 or linear accelerator. The study was planned to estimate the serum protein level at two time periods: (1) at the time of diagnosis (pre-therapy) and (2) at 2 months after the therapy (post-therapy). In this study, only 36 out of 60 HNSCC cases completed the course, and their blood samples were collected two months after receiving the therapy; the remaining cases dropped out of the study.
Two-milliliter venous blood was collected from each individual in aseptic conditions and was allowed to settle for 30 minutes at room temperature. Buffy coat was removed and was centrifuged at 3,000 rpm for 10 minutes. Serum was collected as the supernatant and was then stored at –80°C.
2. Quantitative analysis of serum p38α, β, γ, and δ in the study group: by surface plasmon resonance technology
BIAcore 3000 (Wipro GE Healthcare, Uppasala, Sweden) was used for all label-free real-time monitoring of target bimolecular interactions. Mouse antihuman p38α, p38γ, and p38δ monoclonal IgG and goat antihuman p38β polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, CA) were separately immobilized on different flow cells of CM5 sensor chip via amine coupling kit (Wipro GE Healthcare). Antibodies were diluted to 100 μg/mL in 10 mM sodium acetate (pH 5.0) and were injected over the activated chip surface [3].
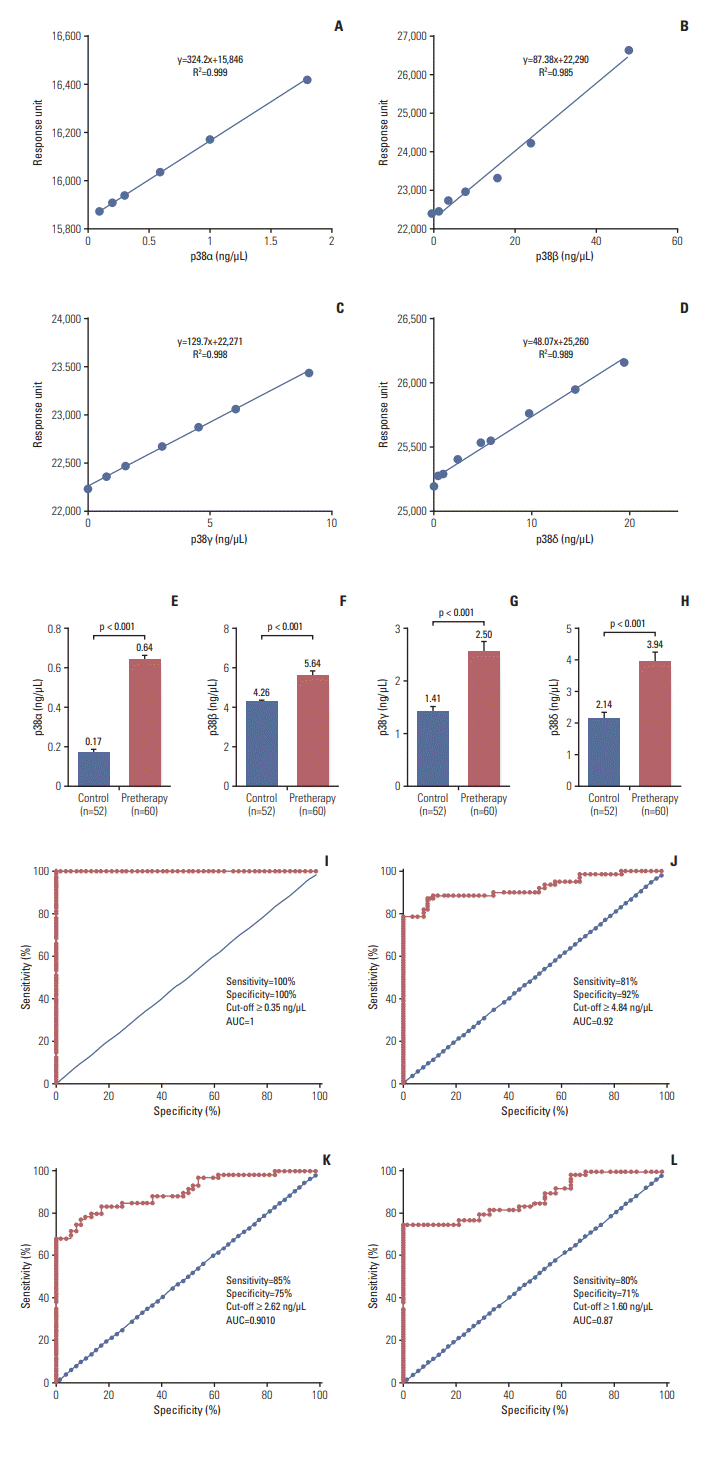
Standard graphs were prepared by passing different known concentrations of pure p38α (0.1, 0.2, 0.3, 0.6, 1.0, and 1.8 ng/μL), p38β (0, 1.6, 4.0, 8.0, 16, 24, and 48 ng/μL), p38γ (0.30, 0.76, 1.52, 3.05, 4.58, 6.1, and 9.17 ng/μL), and p38δ (0.48, 0.97, 2.42, 4.8, 5.8, 9.7, 14.5, and 19.4 ng/μL) over respective antibodies on sensor chip and obtained response unit (RU) .
Serum samples were diluted (1.4:98.6 μL) with HBS-EP buffer and were allowed to run over immobilized antibodies. The concentrations of p38α, β, γ, and δ proteins of all study groups were determined from respective standard curves.
3. Design of peptide using molecular docking
p38α is a well-studied and peptide was earlier designed in our laboratory based on ATP and DFG binding sites for the same from our lab [4,5]. This study designed a peptide based on p38δ, and we compared it with other isoforms.
1) Glide: Grid-based Ligand Docking with Energetics
Glide is a computer-based program employed for screening and docking of small chemical and peptidomimetic libraries acting as ligand against a target protein. The program explores the appropriate molecular interactions between these ligands and the target protein. The first line of hierarchical filtering screened the ligand on the basis of their topological fit with the receptor grid and was followed by evaluating for compatible interactions between ligands and corresponding receptor/protein. Ligand poses that qualify for first-line of screening/filtering were then subjected to computation and minimization of grid approximation to calculate nonbonded interaction energies between ligand and receptor using all-atom optimized potentials for liquid simulations parameters. These poses were scored using Glide Score scoring function [6].
2) Preparation of Tetra-peptides library
Tetrapeptides library with 1.6 lacs peptides was generated using a python script, which was then parsed to Pymol molecular modeling and visualization software. Ligand docking in Glide required the preparation of ligand, which was done through Ligprep wizard of Glide docking tool. The Ligprep wizard generated a maximum of 32 stereoisomers per ligand. Furthermore, we used Epik module in generating possible states within pH 7.0±2.0.
3) Protein preparation
Crystal structures of p38α (PDB: 1A9U), p38β (PDB: 3GP0), p38γ (PDB: 1CM8), and p38δ (PDB: 4EYM) were downloaded from the protein data bank (https://www.rcsb.org). Prior to docking, the heteroatom-like cocrystallized inhibitor and water molecules were removed from all crystal structures. Missing 1CM8 and 3GP0 residues were built using a prime module. All protein structures were prepared through the protein preparation wizard of Glide, which sets bond orders, appends hydrogen, and creates disulfide bonds. Lastly, the prepared proteins were minimized by OPLS_2005 force field using Impref minimization wizard of Glide.
4) Screening and docking of peptide library
After protein and ligand preparation, computational screening of the peptide library was executed against p38δ as the target protein. To screen and dock the peptides, a grid of 23 Å was elucidated around the ATP-binding site by taking the residues around the 5 Å region of the co-crystallized inhibitor present in the structure. Similarly, for the docking of other p38 MAPK isoforms, the grids were prepared by targeting the ATP-binding region. The peptide library was screened through high-throughput virtual screening and was followed by standard precision modes to filter the top hits. Finally, peptide docking was performed using extra precision mode. The scheme of the tetrapeptide was screened and docked against the p38 MAPK.
4. Synthesis of peptide
The peptide was synthesized through SPPS in PS3 peptide synthesizer using Fmoc and Wang resin chemistry. The resin used was a Wang resin, and the solvent was dry distilled N,N-dimethylformamide [7]. The synthesized peptides were purified and were used for other biochemical studies.
5. Binding studies of peptide with p38δ by surface plasmon resonance
Anti‒glutathione-S-transferase (GST) antibody was immobilized on the CM5 sensor chip at 25°C in BIAcore-3000 apparatus on flow cells 1 and 2 using an amine coupling kit. Then, pure GST (Wipro GE Healthcare) and pure recombinant GST-tag p38δ protein were passed over flow cells 1 and 2, respectively.
The pure protein p38α was immobilized on Ni-NTA chip.
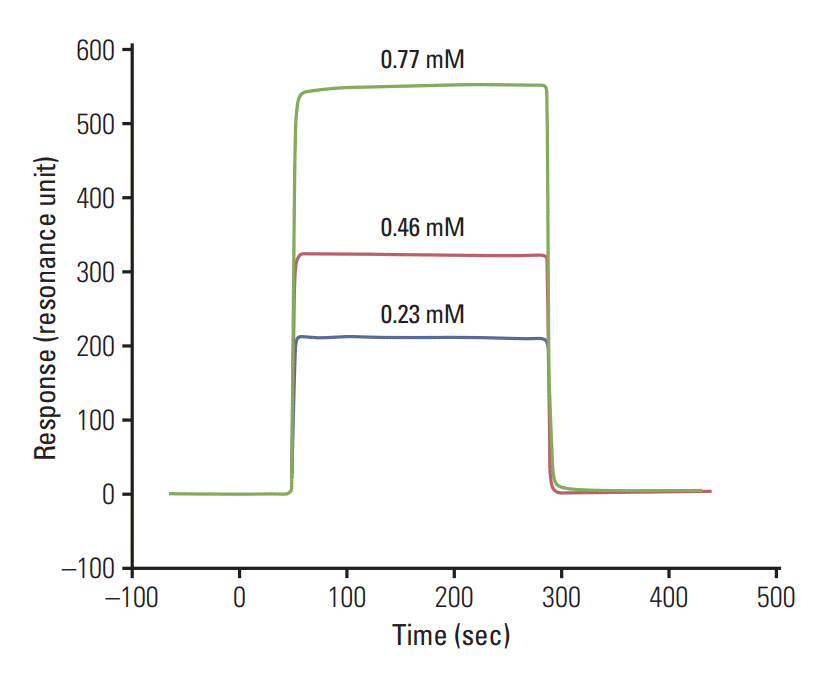
The binding analysis of the synthesized peptide WFYH was performed by passing three different concentrations (0.23, 0.46, and 0.77 mM) of peptide prepared in HBS-EP buffer over the immobilized proteins. The sensorgram was recorded, and the association (KA) and the dissociation constants (KD) were calculated by BIA evaluation 3.0 software.
6. Percentage inhibition assay by enzyme-linked immunosorbent assay
The activity of p38δ was measured using activating transcription factor 2 (ATF-2) as a substrate. Microtiter plates were coated with 10 μL/mL ATF-2 solution (Sigma-Aldrich, St. Louis, MO) at 4°C, and 12 μg purified p38δ protein in kinase buffer (50 mM Tris, pH 7.5, 10 mM MgCl2, 10 mM β-glycerophosphate, 100 μg/mL bovine serum albumin, 1 mM dithiothreitol, 0.1 mM Na3VO4, 100 μM ATP) was added to the wells and was incubated for 1 hour at 37°C. The kinase mixture without the p38δ protein was used as a blank. The plates were incubated with antiphospho-ATF-2 antibody (1:400, Biovision Inc., San Francisco, CA) for 1 hour at 37°C, which interacted with the phosphorylated ATF-2, and were subsequently incubated with alkaline phosphatase conjugated goat antirabbit IgG (1:4,000, Chemicon, Temecula, CA) for 1 hour at 37°C. Finally, the chromogenic substrate solution 4-nitrophenyl phosphate in 0.1 M Tris-HCl (pH 8.1, 0.01% MgCl2, Cayman Chemical Company, Ann Arbor, MI) was added for 90 minutes at 37°C. The reaction was halted by adding 100 μL of 3 N NaOH and measured at 405 nm using enzyme-linked immunosorbent assay (ELISA) reader (BioTek Instruments, Winooski, VT).
The pure p38δ protein was incubated with kinase buffer, with and without the test peptide. The decline in absorbance of yellow product nitrophenolate formed after primary and secondary antibody incubation was measured at 405 nm. The protein in kinase buffer without peptide was used as blank. Its absorbance was considered as 100% activity and was used to calculated percentage inhibition of each peptide. Same experiment was performed with p38α protein.
7. Statistical analysis
For statistical analysis, GraphPad Prism ver. 5.0 (GraphPad Software, San Diego, CA) was used. Descriptive analysis was done for all variables and with percentages or mean (95% confidence interval) as suitable. Student unpaired t test for comparison of two categories was performed for baseline comparison between HNSCC and control population. Receiver operating characteristic (ROC) curve was obtained by plotting sensitivity versus specificity, which is required to obtain cut-off values for given proteins in the serum. Statistical significance was considered with p-value of < 0.05.
Results
1. Demographic data of study groups
The demographic data of HNSCC cases and healthy subjects are described in Table 1. The majority of cases in both groups were males. Furthermore, majority of the cases were active tobacco chewers or smokers. Most of the cases were in their advanced stage (III+IV) disease with histopathologic type, moderately differentiated squamous cell carcinoma.
2. Quantitative analysis of serum p38α, β, γ, and δ in the study group
The binding of the proteins were in the linear range, as shown in the standard curve (Fig. 1A-D). The mean concentrations of p38α, β, γ, and δ of the cases at the time of diagnosis were 0.64, 5.64, 2.5, and 3.94 ng/μL and were 0.17, 4.26, 1.41, and 2.14 ng/μL for the controls, respectively (Fig. 1E-H). The mean concentration of all protein isoforms was compared with the demographic data of the cases (Table 2).
ROC curves for each protein were generated between controls and cases. Based on the surface plasmon resonance (SPR) data, the area under ROC curves was calculated to measure its utility for detection of HNSCC. Based on our data, the obtained cut-off values for p38α, β, γ, and δ were ≥ 0.35, ≥ 4.84, ≥ 2.62, and ≥ 1.60 ng/μL, respectively. For p38α, area under the curve (AUC) yielded sensitivity and specificity of 100% to detect HNSCC. The threshold value yielded a sensitivity/specificity of 81%/92% for p38β, 75%/85% for p38γ, and 71%/80% for p38δ in detecting HNSCC cases (Fig. 1I-L).
Hence, it revealed that the diagnostic efficiency of p38α in detecting HNSCC was higher as compared to other p38 MAPK isoforms and was followed by p38β, δ, and γ.
3. Estimation of expression level of p38MAPK protein after therapy
During clinical assessment, only 36 out of 60 cases underwent treatment. Among these 36 cases, only 31 cases were clinically responders to therapy and demonstrated significant decrease in proteins levels (p38α, β, and δ) after therapy while p38γ concentrations remained unchanged. The remaining five cases were clinically nonresponders and showed no changes in protein concentration of all p38 MAPK (Table 3). The cases were reevaluated by clinicians after completion of therapy, as well as by endoscopy whenever required by radiological examination.
4. Designing of peptide inhibitor using molecular docking
Tetrapeptide library of 1.6 lacs tetrapeptides were screened and docked using GLIDE program (Schrödinger, LLC, New York, NY) against p38δMAPK. Based on the Glide Score, the top 15% of the tetrapeptides were finally docked using extra precision mode of the program; the top ten tetrapeptides were then obtained (S1 Table) and evaluated for postdocking analysis, as well as their selectivity against other p38 MAPK isoforms (Table 4). Based on our docking experiments, the highest docking score computed for the WFYH tetrapeptide (Glide Score) was –12.44. The corresponding Emodel value for this tetrapeptide was also highest among other top ten tetrapeptides and was computed to be –151.70.
5. Post-docking analysis
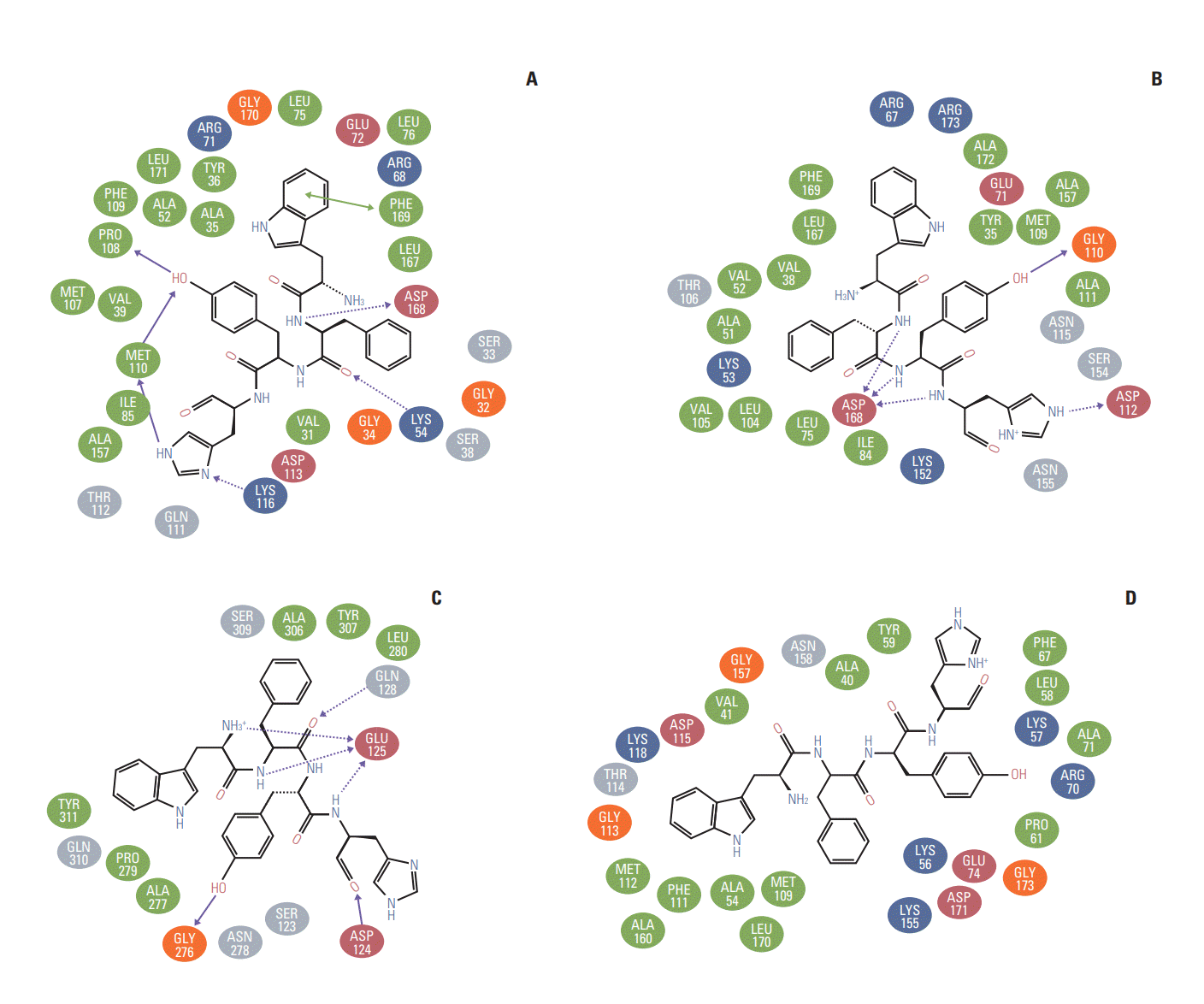
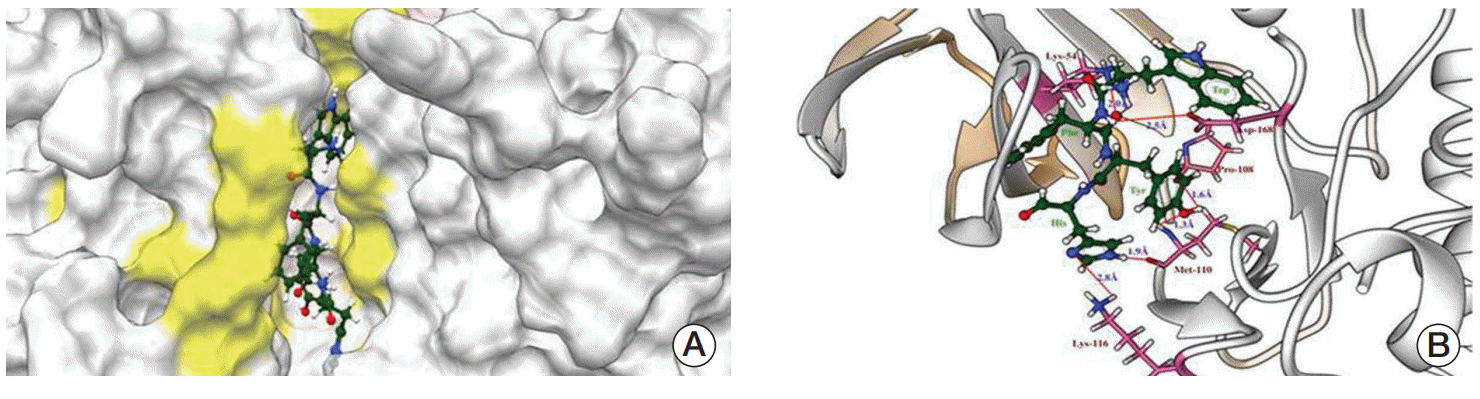
The molecular docking experiments were further validated using postdocking analysis programs: X-score and Lig interaction diagram. The WFYH tetrapeptide interacted with six hydrogen bonds, and it also formed π-π interaction with Phe-169 (Fig. 2A-D), which is a key residue in p38 MAPK family members. To determine the selectivity of WFYH tetra peptide, we further docked it with other p38 MAPK isoforms at the ATP binding site. Surface structure of p38δ illustrates a docked WFYH tetra peptide at its ATP-binding site (Fig. 3A). Ribbon diagram showing Lys-54, Pro-108, Met-110, Lys-116, and Asp-168 residues of p38δ hydrogen bonding with WFYH tetra peptide (Fig. 3B).
6. Binding studies of peptide with p38δ by SPR
The RU values of immobilized anti-GST antibody on flow cells 1 and 2 were 7,004.4 and 7,174.2, respectively (S2 Fig.). The recombinant GST protein immobilized on flow cell 1 showed RU of 24,545.0, whereas the RU observed for p38δ pure protein immobilized on flow cell 2 was 24,666.3. On the other hand, the RU observed for p38α immobilization on Ni-NTA sensor chip was 18,639.
7. Binding analysis of WFYH peptide with p38δ
The analysis of peptide binding was done by passing three different concentrations. The calculated KD analyzed by BIA evaluation 3.0 software value of the WFYH peptide with p38δwas (7.04±0.1)×10–10 M (Fig. 4), whereas no binding was observed in the case of WFYH with p38α.
Discussion
The expression of p38 MAPK isoforms varies in different cell types. Among four isoforms, p38α has been well studied in HNSCC and in other cancers. Elevated serum p38α level in HNSCC was reported earlier from our laboratory [3]. This study aims to explore the expression levels of all p38 MAPK isoforms in serum of HNSCC patients and to identify specific protein markers for early detection of such disease. In our study, we found a significant decrease in the expression patterns of p38α and p38δ (p < 0.001) after therapy in clinically responder patient group, thus clearly suggesting its importance in the diagnosis and prognosis of HNSCC, as it can be a good therapeutic target as well. In contrast, the expression level of p38γ was also associated with HNSCC; however, after therapy, its concentration in serum remained unchanged.
Different tissue and cellular expression status of p38 MAPKs suggest their association with distinct signaling pathways; thus, targeting one of the four isoforms, p38δ can be beneficial in alleviating associated symptoms. However, the candidate ligand must be specific against only one target isoform and should not inhibit the other isoforms of the family.
Studies have shown that although the members of p38 MAPKs family share similarities, yet they can be categorized into two subsets on the basis of substrate phosphorylation and activation [8] and comparative sequence similarities between the subsets and expression profile. This characterization can be well understood by the inhibition pattern among these isoforms, wherein one subset (p38α and β) is sensitive to pyridinyl imidazole drugs, whereas the other subset (p38γ and δ) is resistant to these drugs [9]. Thus, to study the specificity of the WFYH tetrapeptide, we also docked it and the other top nine tetrapeptides against the ATP-binding sites of p38α, β, and γ. Based on the docking and postdocking analysis, the WFYH tetrapeptide computed with p38 isoforms and showed the best Glide Scores with p38δ, hence indicating poor binding potentials of WFYH with other isoforms (Table 4).
p38 MAPKs exhibit two states, namely active and inactive. The transformation of these isoforms, from inactive to active configuration, is due to the conformational rearrangements within the protein structures, which are induced by dual phosphorylation at Thr and Tyr moieties localized in the activation loop (residues 168-186). In p38δ, Thr-180 and Tyr-182 are phosphorylated, which are coordinated by R-71, R-149, R-177, R-186, and R-189. Similarly, in p38α phosphorylated Thr-180 and Tyr-182 are coordinated by R-67, R-70, R-171, R-189, and H-228; in p38β, phosphorylated Thr-183 and Tyr-185 are coordinated by K-69, R-73, R-152, R-176, R-189, and R192 [10]; and in p38γ, phosphorylated Thr-183 and Tyr-185 are coordinated with K-69, R-70, R-73, R-152, R-176, R-189, and R-192 [11]. Ligands interacting with these coordinating residues will disrupt the structural conformation of active MAPKs. Postdocking analysis of docked WFYH to p38 MAPKs isoforms demonstrated no interaction between WFYH and any of the coordinating residues involved in the stabilization of p38 MAPKs (Fig. 2A-D). In p38 MAPK isoforms, the Phe-169 in the activation loop is a pivotal residue, which shuttles in different conformations in active and inactive forms. Postdocking analysis shows that WFYH tetrapeptide does not interact with Phe-169 of p38β and γ; however, it forms a hydrophobic interaction with Phe-169 of p38α and π-π interaction with Phe-169 of p38δ. π-π interactions are strong noncovalent forces, which play crucial role in maintaining protein structures and in ligand-protein interactions. These forces are well suited for biological interactions encountering a strong hydrophobic environment or when the interacting residue is deeply embedded in the protein [12]. Formation of π-π interaction between the benzene ring of Trp of WFYH and Phe-169 thus confers a strong interacting force, which contributes to enhanced binding affinity of WFYH to p38δ; this interaction also locks the movement of Phe-169.
As there are more than 50% of sequence similarities between p38α and δ, analyzing the interactions between WFYH and these residues can be used to determine both the efficacy and comparative specificity of p38δ, with respect to p38α. The WFYH docked to ATP site of p38δ forms hydrogen bonds with Lys-54, Met-110 localized in the hinge region, and Asp-168 of DFG motif; Pro-108 and Lys-116, on the other hand, when docked to p38α, forms hydrogen bonds with Gly-110, Asp-112, and Asp-168, along with other 14 hydrophobic interactions (Fig. 2B). Multiple studies on different inhibitors exhibiting higher specificity for p38α share some common hydrogen bond forming residues such as Lys-53, Val-105, Leu-108, Met-109, Gly-110, and Asp-168. Most of the p38α inhibitors also interact with Thr-106 and Tyr-35. This data clearly suggests that the WFYH exhibits lower binding affinity for p38α, as it shares hydrogen bonds with only two residues that are commonly shared by other p38α inhibitors. Furthermore, various studies have shown that specific inhibition of p38α by inhibitors requires hydrogen bond formation with Met-109 and Gly-110. This interaction induces a peptide flip of both Met-109 and Gly-110, which contributes to p38α inhibitor selectivity [13-15]. The tetrapeptide WFYH forms hydrogen bond with Gly-110, but not with Met-109, against p38α, thus indicating poor selectivity.
Comparative analysis of WFYH docking with p38α, β, γ, and δ demonstrates that the tetrapeptide WFYH demonstrated good binding affinity for p38δ, in contrast to other isoforms, as observed by comparative lower Glide Scores and differences in the interacting residues.
The obtained KD value by SPR method and % inhibition values determined through ELISA showed that WFYH can be considered as an effective p38δ inhibitor. The specificity of the peptide inhibitor was also confirmed by performing inhibition of pure p38δ. A reduction in phosphorylation of ATF-2 was also observed in kinetic assays involving pure p38δ protein incubated with peptide. Hence, p38δ MAPK inhibitor can be a potential therapeutic agent for HNSCC.
In conclusion, among p38 MAPK isoforms, p38α was found to be associated with overall stage specific and prognosis of HNSCC, and higher p38α levels were associated with disease condition. Based on the SPR data, the AUC was calculated to measure the utility of each protein as potential markers for HNSCC. The obtained ROC curves also revealed that higher values of marker can be used in detecting disease and differentiating cases from healthy controls. Higher levels of proteins were associated with disease conditions. These results provide information regarding p38 MAPK isoforms expression pattern in HNSCC and also report that p38δ can be a very promising protein marker, as well as a therapeutic target.
 XML Download
XML Download