

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC) is the third most common cancer diagnosis, and is the leading cause of cancer-related death worldwide [1]. However, because existing therapies can be toxic, more specific therapeutic regimens, such as targeted agents, have been sought to improve the outcomes and quality of life of patients with CRC [2]. To do so, many studies are underway to discover new targets for targeted therapy through genome analysis.
Fusion transcripts (FTs) were recently reported to produce feasible drug targets, which implies that detection of FTs could be a plausible diagnostic and/or treatment strategy for some malignancies [3-6]. Characterization of specific FTs in cancers has led to the identification of several successful therapeutic targets, such as BCR–ABL1 [7]. FTs are a class of genetic alterations that result from chromosomal rearrangements [8]. FTs that result in tumorigenesis are potential drug targets in several cancers [9]. Many tyrosine kinase rearrangements occur in epithelial cancers, including anaplastic lymphoma kinase (ALK), proto-oncogene tyrosine-protein kinase ROS (ROS1), ret proto-oncogene (RET), neurotrophic tyrosine kinase, receptor, type 1 (NTRK1), fibroblast growth factor receptors (FGFR), AXL receptor tyrosine kinase (AXL), and platelet-derived growth factor receptor, alpha polypeptide (PDGFRA) [10]. Among these tyrosine kinase fusions transcripts, the echinoderm microtubule-associated proteinlike 4 ALK (EML4–ALK) fusion, which was discovered in non-small-cell lung cancer (NSCLC) [11], has been targeted by crizotinib for NSCLCs that harbor the EML4–ALK fusion gene [12].
Of the many FTs that have been discovered in CRC thus far, almost all, including VTI1A–TCF7L2 [13], PTPRK–RSPO3 [14], and TCF7L2–RP11–57H14.3 [15], affect the Wnt signaling pathway. Many FTs have been discovered in CRC, including AKAP13–PDE8A, COMMD10–AP3S1, SLC39A14–TSPAN15, NCOA3–SPINT1, GRIN2B–CYP4F3, PRMT1–FLT-3LG, and VNN1–AB [16,17]. However, with the exception of VTL1A–TCF7L2 and PTPRK–RSPO3, their functions have not been studied. As with other FTs in diverse tumors, the identification of FTs in CRC could provide new insights about this disease.
Here, we conducted paired-end RNA sequencing to discover novel FTs in CRC cell lines. NFATC3–PLA2G15 FT was also detected in CRC tumor tissue. We conducted experiments aimed at a functional study of the NFATC3–PLA2G15 FT. We observed that NFATC3–PLA2G15 FT was involved in the regulation of invasion, anchorage-independent features, and cell proliferation in CRC cell lines.
Materials and Methods
1. Cell lines and culture
Human CRC cell lines (colo-201, colo-205, colo-320, HCT-15, HCT-116, HT-29, Lovo, LS174T, SNU-61, SNU-70, SNU-81, SNU-175, SNU-254, SNU-283, SNU-407, SNU-977, SNU-1033, SNU-1181, SNU-1235, SNU-1411, SNU-1544, SNU-1684, SNU-C1, SNU-C2A, SNU-C4, SNU-C5, SW-403, and SW-480) were purchased from the Korean Cell Line Bank [18]. All cell lines were cultured in RPMI1640 (Hyclone Laboratories, Ind., Logan, UT), supplemented with 10% fetal bovine serum, at 37°C in humidified 5% CO2.
2. Paired-end RNA sequencing and fusion analysis
Total RNA was extracted from each cell line using an RNeasy mini kit (Qiagen, Valencia, CA). The mRNA was isolated from 1 μg of total RNA using the NEBNext Poly mRNA Magnetic Isolation Module. The cDNA library was prepared from mRNA using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) and sequenced using the Illumina HiSeq 2500 platform (2×150 bp).
Next-generation sequencing reads were analyzed with three fusion detection tools including TopHat-fusion, ChimeraScan, and FusionMap. TopHat-fusion (v.2.0.9) analysis was performed using Bowtie (v.1.0.1) with fusionsearch option and default parameters. ChimeraScan (v.0.4.5) and FusionMap (2013-07-30) were also performed using default parameters. All of the programs used hg19 (GRCh37) and RefGene as reference genome and gene model, respectively. From all of the results analyzed using the three tools, we obtained fusions with four or more supporting reads for further analysis.
3. Human colon cancer tissues
We obtained 19 human colon adenocarcinoma tissues and matched normal tissues from the tissue bank of Seoul National University Hospital (Seoul, Korea). After resection, the tissues were immediately frozen in liquid nitrogen and stored until needed.
4. Reverse transcriptase-polymerase chain reaction and quantitative real-time polymerase chain reaction
Total RNA was extracted from each cell line for transcriptome sequencing using an RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. The cDNA was synthesized from total RNA (1 μg) using ImProm-II reverse transcriptase (Promega, Madison, WI) and amplified by reverse transcriptase–polymerase chain reaction (RT-PCR) using AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA) with buffer supplied by the manufacturer and gene/fusion junction-specific primers. For quantitative realtime RT-PCR (qRT-PCR), cDNA was amplified using Premix Ex Taq (TaKaRa, Shiga, Japan) with SYBR Green I (Molecular Probes, Eugene, OR) using a StepOnePlus Real-Time PCR system (Applied Biosystems).
5. Sanger sequencing
Sanger-sequencing reactions were conducted in both forward and reverse reactions. NFATC3–PLA2G15 FT junction was amplified using the following forward (F): GTGTGATCCAGCGTCATTT and reverse (R): AGTAGGGCCCGTTTTCATTT primers. Sequencing was carried out by Macrogen Inc. (Seoul, Korea).
6. IGV data analysis
The IGV program (version IGV_2.3.32) was downloaded from https://www.broadinstitute.org/igv/. After loading the BAM file from the RNA-sequencing raw data, the 3′ position of NFATC3 and the 5′ position of PLA2G15 were analyzed.
7. Small interfering RNA knockdown
The siRNAs used in this study, including the control siRNA (si-CONTROL) with scrambled sequences, were synthesized by Genolution Pharmaceuticals, Inc. (Seoul, Korea). The siRNA sequences used against the NFATC3–PLA2G15 FT were 5′-GAUGAUGUCCCUGGUGAUUUU-3’ (sense) and 5′-AAUCACCAGGGACAUCAUCUU-3′ (antisense), which targeted the FT breakpoint. Each cell line was treated with siRNA (50 nM) for 48 hours. Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used for transfection, according to manufacturer’s instructions.
8. Soft agar assay
Noble agar (Difco, HR, India), which was prepared by combining 1.5 mL of 0.5% agar in RPMI1640 with 20% fetal bovine serum (FBS), was solidified in the bottom of each well of a 6-well plate. On the next day, si_NFATC3-PLA2G15 fusion and control transfected cells (1×104) in RPMI1640 with 20% FBS media were mixed with 0.4% agarose and overlaid onto the bottom agar. The plates were then incubated in a 5% CO2 incubator at 37°C. After 3 weeks, colonies were counted under a microscope and numbers of colonies from triplicate wells were averaged. Colonies were imaged using a microscope equipped with a digital camera.
9. Invasion assay
3×105 colo-320 cells were stably transfected with si-control and si-NFATC3-PLA2G15 and 3×104 cells were seeded on the Matrigel-coated membrane matrix (BD Biosciences, San Jose, CA) insert well according to manufacturer’s instructions after 48 hours. Media containing 20% FBS was added as a chemoattractant to the wells of the Matrigel invasion chamber for 24 hours. The following day, the cells were fixed for 10 minutes in 3.7% paraformaldehyde and the insert was washed with phosphate-buffered saline (PBS). Crystal violet (0.1%) was added to the insert for 10 minutes and washed twice with PBS, and then with water. A cotton swab was used to remove any non-invading cells and the insert was washed again. The number of invading cells was imaged using a microscope equipped with a digital camera.
10. Cell proliferation assay
Colo-320 cells were plated in completed growth media in 60 mm dishes (7×105 cells per dish) and treated with si_NFATC3-PLA2G15 and si_control on the following day. Cell numbers were counted using a hemocytometer for 4 days in a time-dependent manner. Three independent experiments were performed, and the results averaged.
11. Western blotting
Cultured cells were washed with ice-cold PBS and lysed with lysis buffer (50 mM Tris-HCl [pH 7.5], 1% NP-40, 0.1% sodium deoxycholate, 150 mM NaCl, 50 mM NaF, 1 mM sodium pyrophosphate, 1 mM EDTA, and protease/phosphatase inhibitors). Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used to resolve 20 μg of total protein. The resolved proteins were then transferred to nitrocellulose membranes. After blocking with 1% skim milk and 1% bovine serum albumin/Tris-buffered saline with Tween 20, the membranes were incubated with primary antibodies at 4°C overnight. Antibody against cyclin D were purchased from Cell Signaling Technology (Beverley, MA). Antibodies against vimentin, E-cadherin, cyclin E, and p27 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). An anti-β-actin antibody was acquired from Sigma-Aldrich (St. Louis, MO).
Results
1. Paired-end sequencing and identification of FTs
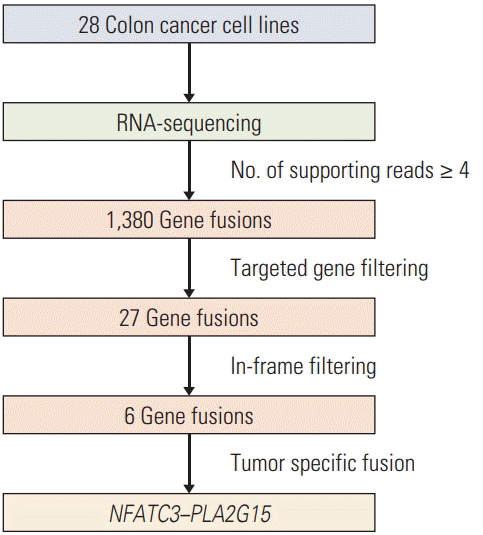
To discover novel gene fusions in CRC, we performed paired-end RNA sequencing using 28 CRC cell lines. Average depth of RNA-sequencing data was 154× and median depth was 146×. Statistics of each RNA-sequencing data was indicated in S1 and S2 Table. The list of FT candidates was produced by using the TopHat-fusion, ChimeraScan, and FusionMap tools, for a total of 1,380 candidates with at least four spanning read counts at each of the tools.
We followed a targeted approach to filter the 1,380 gene fusions that contained Wnt signaling pathway-related genes and kinase domains for target gene selection. First, we obtained the list of Wnt signaling pathway-related genes from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway because activation of the Wnt signaling pathway is a critical event in the development of CRC. Second, we composed a list of the genes from DAVID that included a kinase domain, which could be a biomarker for a therapeutic target.
In the targeted filtering step, 139 WNT-related genes and 11 genes with kinase domains were selected as FT candidates. After targeted filtering, 27 FTs remained. We removed 21 gene fusions because they did not have in-frame sequences where the two genes were fused (Fig. 1). Finally, four intra-chromosomal FTs (PPP2CA–SKP1, CTNNBIP1–CLSTN1, AKAP13–PDE8A, and NFATC3–PLA2G15) and two inter–chromosomal FTs (FZD2–RPS24 and KRT8–PKM2) remained (Table 1).
2. Validation of FTs in cell lines and tumor tissues
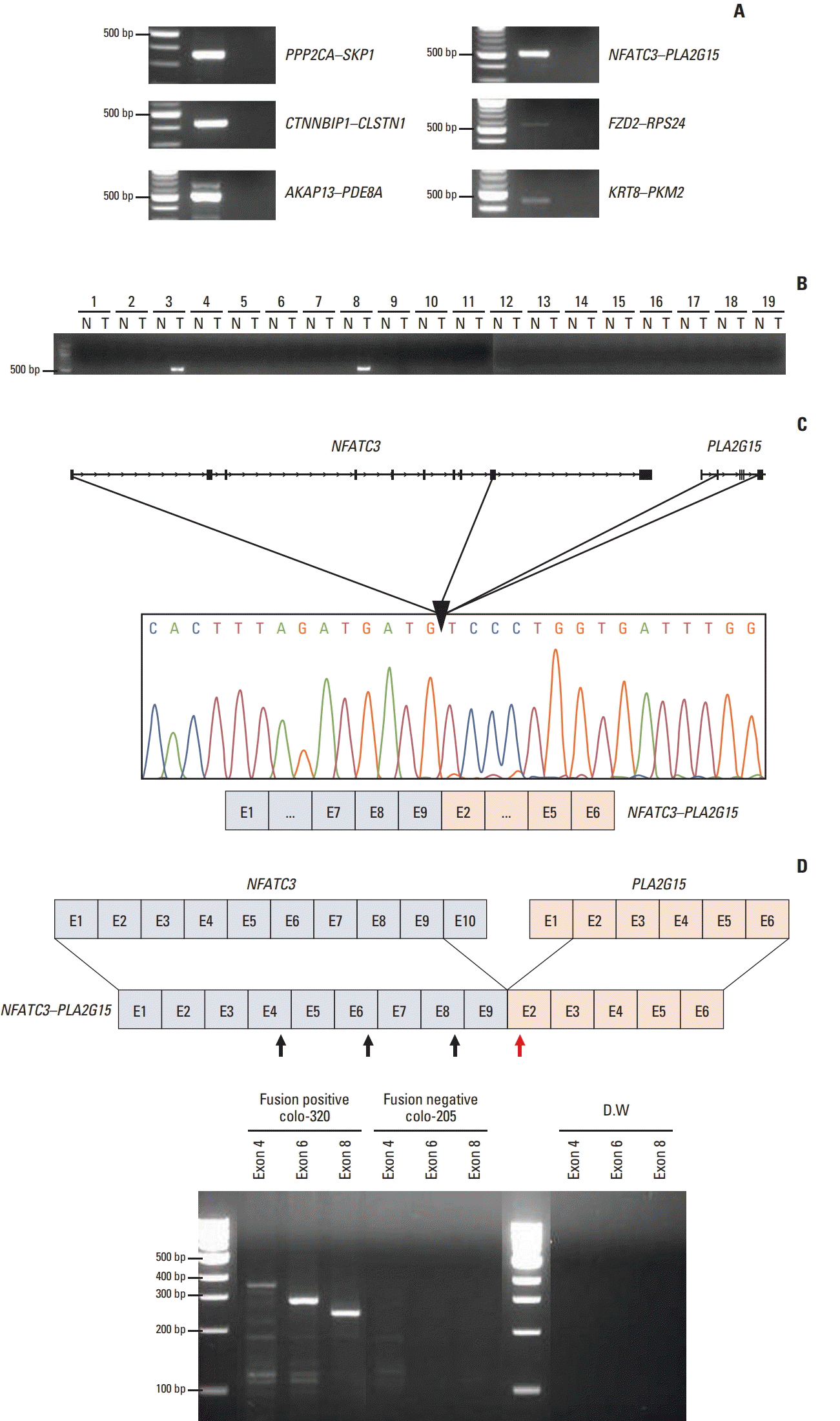
In the validation step, each of six FTs—PPP2CA–SKP1, CTNNBIP1–CLSTN1, AKAP13–PDE8A, NFATC3–PLA2G15, FZD2–RPS24, and KRT8–PKM2—were detected in HCT-15, SNU-81, SW-480, colo-320, HCT-15, and SNU-1235, respectively (Fig. 2A). We next examined the frequency of the FTs in tumor tissues and matched normal tissues. Only NFATC3–PLA2G15 FT was detected in two out of 19 (10.5%) tumor samples, but not in matched normal tissues, which implies that NFATC3–PLA2G15 is a tumor-specific transcript (Fig. 2B). Moreover, age, sex, histology, primary site, microsatellite stable/microsatellite instable status, or tumor stage of the CRC patients did not show a significant correlation (data not shown). The other FTs were not detected in the tumor tissue (data not shown). Therefore, we focused on the NFATC3–PLA2G15 FT for further study.
The NFATC3–PLA2G15 FT consisted of exons 1–9 of NFATC3 and exons 2–6 of PLA2G15; fusion junctions of each transcript were confirmed by Sanger sequencing (Fig. 2C). However, no genomic-level changes were detected by long-range PCR (data not shown). We designed a set of forward PCR primers that targeted NFATC3 exons 4, 6, and 8, and reverse primers for PLA2G15 exon 2 to detect the FT directly by reverse transcriptase‒polymerase chain reaction (Fig. 2D). We confirmed the presence of NFATC3–PLA2G15 FT in colo-320 cells, but not in the fusion-negative colo-205 cells.
3. NFATC3–PLA2G15 FT regulates EMT
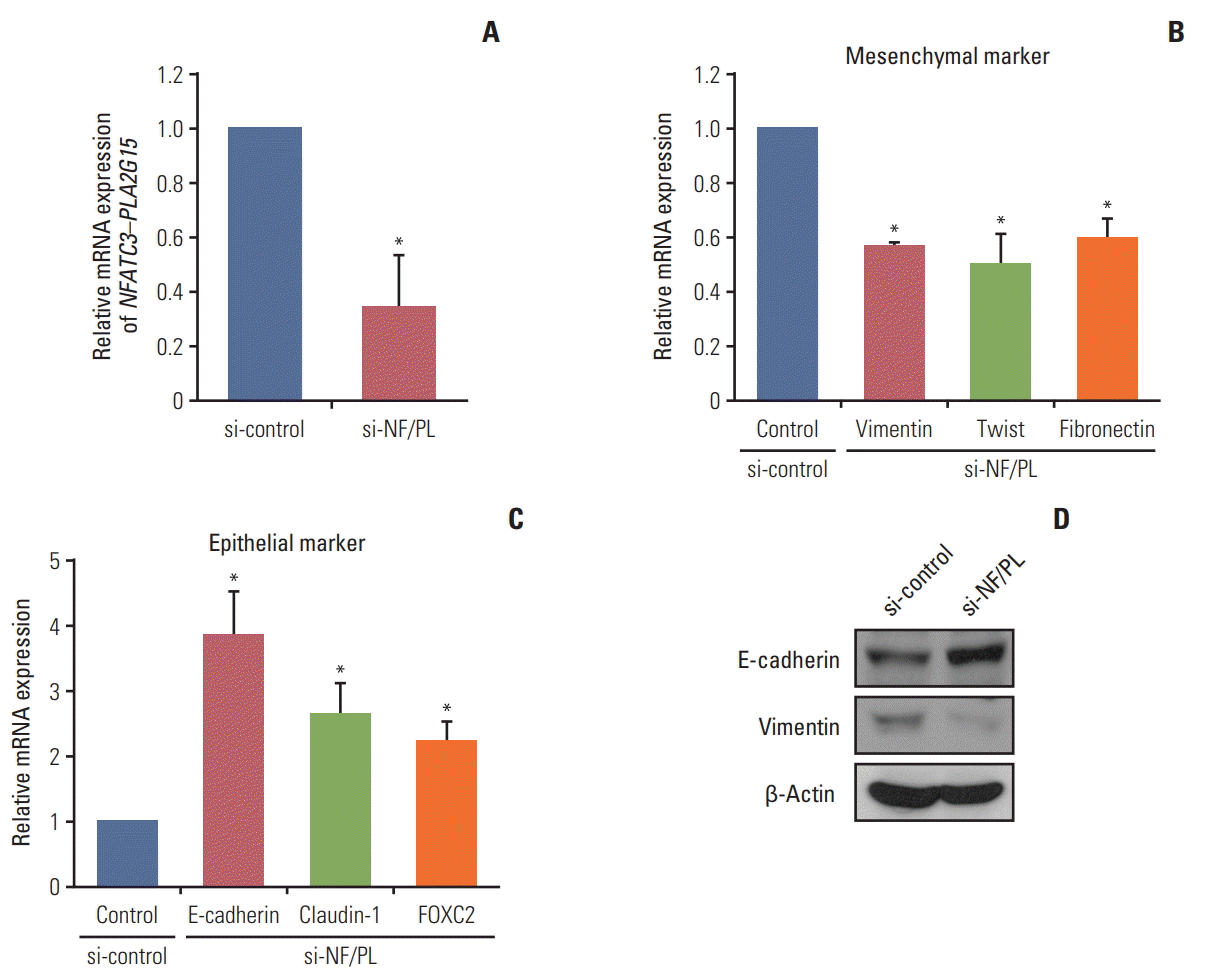
To investigate the functional consequences of inhibiting the NFATC3–PLA2G15 FT, we designed a customized siRNA to target the fusion junction site for a knock-down NFATC3–PLA2G15 FT (NFATC3–PLA2G15-KO). Reportedly, NFATC promotes epithelial–mesenchymal transition (EMT) through upregulation of Src expression in mouse embryonic stem cells [19], whereas silencing NFATC inhibited transforming growth factor β–mediated EMT [20]. Vimentin and E-cadherin are well characterized and have specific roles in mesenchymal and epithelial cellular states, respectively. We therefore screened mRNA expression levels of vimentin and E-cadherin in 28 CRC cell lines using qRT-PCR. We discovered that vimentin mRNA expression levels were the highest and E-cadherin levels were the lowest in the colo-320 cell line that harbored the NFATC3–PLA2G15 FT compared with the other 27 CRC cell lines (data not shown). Accordingly, we hypothesized that NFATC3–PLA2G15 could regulate EMT-related genes. Hence, we used qRT-PCR to investigate mRNA expression of epithelial and mesenchymal markers after siRNA treatments. Following treatment with the NFATC3–PLA2G15-specific siRNAs (Fig. 3A), inhibition of the FT expression also resulted in decreased mesenchymal markers—specifically, vimentin, twist, and fibronectin (Fig. 3B); and increased epithelial markers—specifically, E-cadherin, claudin-1, and FOXC2 (Fig. 3C). Western blot showed vimentin expression was decreased, and E-cadherin expression increased, in siRNA-treated cells (Fig. 3D). This result suggests that the NFATC3–PLA2G15 regulates EMT- and MET-related genes.
4. NFATC3–PLA2G15-KO reduced cell invasion, colony formation, and cell proliferation
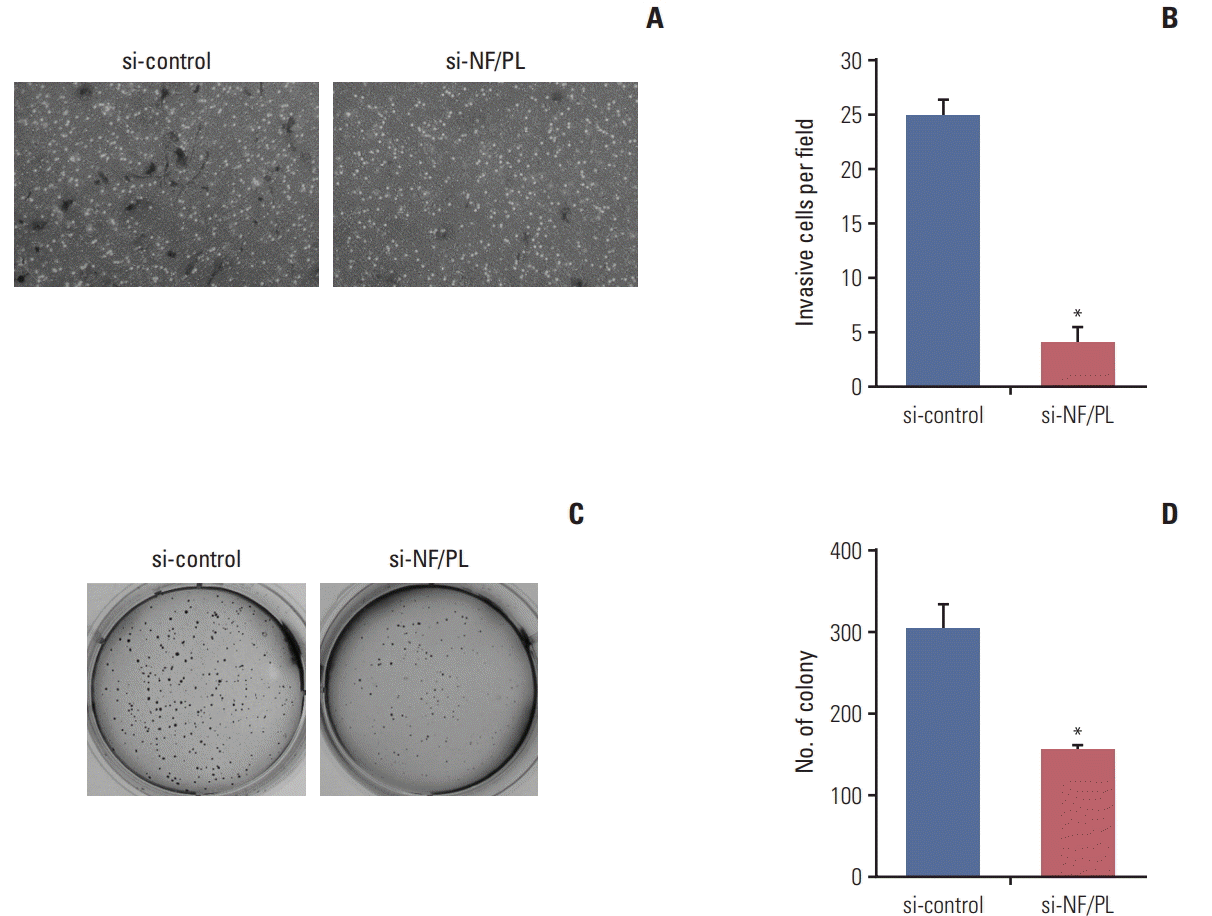
As we observed that NFATC3–PLA2G15 FT could regulate the EMT-related genes, we investigated how NFATC3–PLA2G15-KO affected invasion ability in the cell lines. A Matrigel invasion assay demonstrated that the number of invading cells, NFATC3–PLA2G15-KO cells, was decreased compared with control cells (Fig. 4A and B).
We performed a soft agar assay to investigating the effect of NFATC3–PLA2G15-KO on colony-formation ability in the cell lines. Using fusion junction-specific siRNA, we confirmed knockdown of NFATC3–PLA2G15 mRNA by qRT-PCR. We then observed that colony-formation was suppressed in NFATC3–PLA2G15-KO cells compared with cells treated with control siRNA (Fig. 4C and D). This result showed that NFATC3–PLA2G15 affected cell invasion and colony formation abilities.
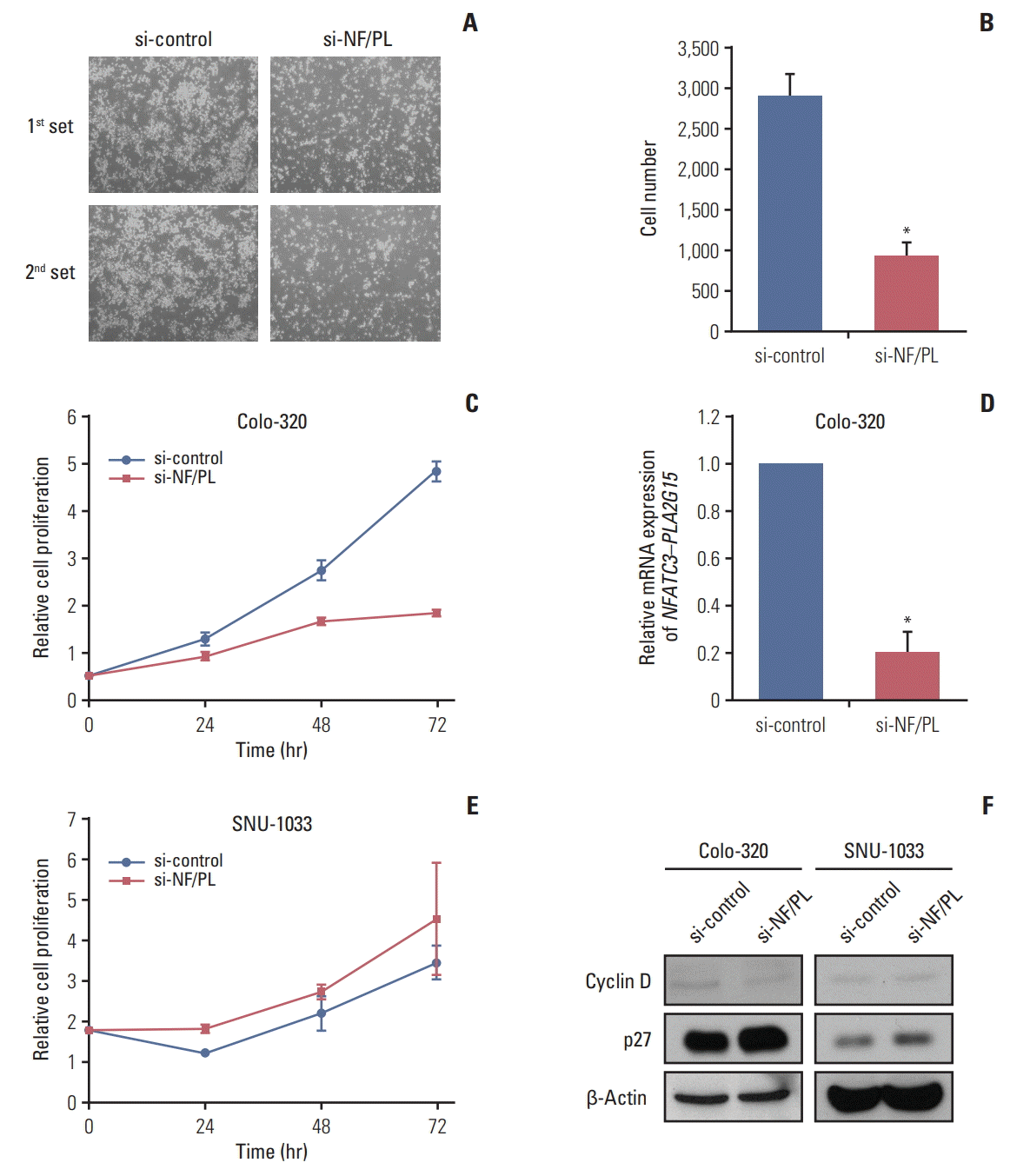
We also conducted a cell-counting assay to investigate the correlation between the NFATC3–PLA2G15 and cell proliferation after siRNA treatment in the colo-320 fusion-positive and SNU-1033 fusion-negative cell lines.
During treatment with NFATC3–PLA2G15 siRNA, confluence of colo-320 cells at 48 hours after siRNA transfection was decreased (Fig. 5A and B). After siRNA treatment, proliferation was inhibited in the colo-320 cell line that had expressed NFATC3–PLA2G15 (Fig. 5C); however, no effects were observed in the SNU-1033 cell line (Fig. 5E). mRNA expression of NFATC3–PLA2G15 was decreased in the colo-320 cell line (Fig. 5D). We also observed decreased cyclin D protein expression and increased p27 expression in the colo-320 cell line but not in the SNU-1033 cell line (Fig. 5F). These results suggested that NFATC3–PLA2G15 regulated cell proliferation.
Discussion
In this study, we identified six FTs and observed the function of NFATC3–PLA2G15 FT. Using paired-end RNA sequencing in 28 CRC cell lines, we obtained 1380 fusion candidates with ≥ 4 reads in online fusion detection tools and filtered them for in-frame fusion patterns to obtain six fusion candidates (PPP2CA–SKP1, CTNNBIP1–CLSTN1, AKAP13–PDE8A, NFATC3–PLA2G15, FZD2–RPS24, and KRT8–PKM2). Among them, we focused on the NFATC3–PLA2G15 FT because it showed tumor-specific characteristics. NFATC3–PLA2G15 FT has previously been reported in a normal-karyotype AML having the same fusion junction as we discovered in this study [21]. However, this study is the first to conduct a functional study of this FT. Although fusion break point is different compared with NFATC3–PLA2G15 FT in our paper, it was additionally detected in T-acute lymphoblastic leukemia recently [22]. The transcription factor, nuclear factor of activated T-cells (NFAT), exists as five isoforms (NFAT1, NFAT2, NFAT3, NFAT4, and NFAT5 [23], and is known to be involved in the non-canonical WNT signaling pathway of the immune system and the WNT/calcium pathway [24]. The binding of Ca2+ to calmodulin activates calcineurin, which dephosphorylates and activates the NFAT transcription factors. Dephosphorylated NFAT then translocates to the nucleus where it interacts with diverse transcriptional partners and regulates the transcription of target genes through a nuclear localization signal (NLS) peptide [25]. NFAT is then re-phosphorylated and inactivated by NFAT kinases, including GSK3, CK1, and DYRK1, after which NFAT translocates back to the cytosol through a nuclear export signal (NES) peptide [26]. The phospholipase A2 Group XV (PLA2G15) protein (also known as lysosomal phospholipase A2 [LPLA2]) acts on the membrane to regulate lipid catabolism; it has calcium-independent activity [27]. Phospholipase mediates of intra- and extra-cellular signaling and affects various tumorigenic processes, including cell migration, invasion, proliferation and angiogenesis [28].
We found that NFATC3–PLA2G15 FT includes exon 1-9 of NFATC3 and exon 2-6 of PLA2G15. Thus, this FT has the NFATC3 NLS peptide like NFATC3 full-length gene, but the NES peptide was deleted when the transcript fuses with PLA2G15. In accordance with it, we hypothesized that the NFATC3–PLA2G15 FT could not translocate back to the cytosol because it did not have the complete NES signal peptide. We tried to investigate this mechanism, but failed to make FT construct and FT gene synthesis. Therefore, follow-up study is needed to discover the mechanism. In accord with a prior report indicating that NFATC3 is involved in the regulation of cell proliferation, we observed that the NFATC3–PLA2G15 FT regulated cell proliferation as a driver gene despite the fact we could not reveal the functional mechanisms. Another group observed that NFAT blocker A-285222 inhibited cell proliferation following a reduction in cyclin D expression [29]. Although we did not investigate a relationship between the NFATC3–PLA2G15 FT and any drug, it could potentially serve as a therapeutic target in CRC. In other reports, the fusion genes containing druggable tyrosine kinase (ALK, FGFR2, KIT, NTRK1, NTRK2, RET and PDGFRA) was verified in colorectal cancer cell lines using targeted approach [30]. As this case, the other 5 fusion candidates from our study are also potential drug targets, and should be screened with more paired tumor tissues. Other than 6 FTs identified through targeted gene filtering step, the functional study of the other high-frequency FTs from other filtering methods, will be needed.
In summary, we have identified expression of NFATC3–PLA2G15 FT in CRC cell lines using RNA sequencing, and demonstrated that recurrent in-frame NFATC3–PLA2G15 FT promoted tumor cell proliferation and invasion ability in CRC cell lines.
 XML Download
XML Download