

 PDF
PDF Citation
Citation Print
Print


Introduction
The estrogen receptor (ER) signaling is of vital importance for the development and progression of breast cancer (BCa). In fact, about 70%-80% of postmenopausal female patients with BCa are ER-positive. So these patients benefit a lot from endocrine therapies such as blocking estrogen binding to ER with antiestrogens (i.e., tamoxifen) or inhibition of estrogen synthesis using aromatase inhibitors (AIs; i.e., letrozole and anastrozole). Compared to tamoxifen, letrozole possesses superior efficacy to prolong the disease-free and overall survival, including higher response rates and lower risks of endometrial cancer and stroke [1]. Despite these advantages, however, resistance eventually occurs in a large portion of letrozole-treated patients, thus resulting in BCa relapse with higher metastatic status and poorer prognosis [2]. Evidence from experimental and clinical studies have identified several potential mechanisms coexisting to confer letrozole-resistance, including repression of ER expression, activation of phosphoinositide-3 kinase (PI3K)/AKT/mammalian target of rapamycin pathway [1] and deregulation of epigenetic modification [3]. Nevertheless, occurrence of letrozole-resistance likely involves actions at different levels and remain to be fully established.
Due to a wide spectrum of biological activities, the interferons (IFNs) pathways have been implicated in multiple malignancies. IFNs bind to type I (IFNα/β) and type II (IFNγ) receptors (IFNAR1/2), and then lead to phosphorylation and activation of various downstream pathways including JAK, AKT, STAT1/2, and mitogen-activated protein kinase, thereby enhancing cell proliferation and survival over therapeutic stress [4]. So deregulation of IFN signaling pathway lays a good foundation for cancerous progression. Very recently, by using global gene expression analysis, Ariazi et al. [5] have identified the IFN signaling as one of the most upregulated pathways involved in tamoxifen-resistant BCa cells. Moreover, using estrogen-deprived human BCa cells, Choi et al. [4] have confirmed that targeting IFN response genes, such as IFN-inducible transmembrane protein 1 (IFITM1, a IFN-inducible cell surface protein), could effectively sensitize the endocrine-resistant BCa cells to estrogeninduced cell death [4]. These studies underscore and emphasize the need to better understand the mechanisms by which how these IFN-inducible genes are regulated and exhibit their pleiotropic effects.
The paired-like homeodomain transcription factor 2 (PITX2), acting as the upstream activator of the muscle regulatory factors, plays a decisive role in skeletal muscle development [6]. A growing body of evidence suggest that additional, as yet poorly characterized, cancerous actions of PITX2 are likely to occur. Patients with aberrant methylation of PITX2 promoter show a significantly higher risk of disease progression in non-small-cell lung cancer [7], BCa [8], and prostate cancer [9]. Moreover, deregulated PITX2 expression potentiates chemoresistance in various malignancies, including esophageal squamous cell carcinoma [10], renal cancer [11], and bladder cancer [12]. Of particular interest, in a previous genome-wide chromatin immunoprecipitation sequencing study, PITX2 is found to be among the top 4 upregulated genes represented in tamoxifen-resistant MCF7 cells [13]. Although these findings suggest broad implication of PITX2 in endocrine resistance of BCa, a role for PITX2 in regulating against endocrine therapy in ER-positive BCa cells, if any, has not been investigated.
We show here, for the first time, that interaction between PITX2 and IFNα signaling pathways strongly promotes cell survival and invasiveness upon letrozole treatment, thus conferring letrozole-resistance in BCa cells.
Materials and Methods
1. Patient samples
Female BCa patients, who had received letrozole 2.5 mg daily in neoadjuvant treatment, were recruited from Department of Breast Surgery in Liaoning Cancer Hospital and Institute during June 2015 and September 2017. Patients were subdivided into “Primary” (complete or partial response to letrozole, n=24) and “Recurrent” (stable or progressive disease after letrozole treatment, n=20) groups based on medical image examination. An incisional biopsy was obtained before new therapy. Moreover, adjacent normal breast tissues sampled at least 5 cm from primary tumors were obtained from 12 chemotherapy-naive BCa patients during mastectomy, and were used as controls. The clinical characteristics of BCa patients recruited in the current study was categorized according to the St. Gallen standards [14] and summarized in S1 Table.
2. Real-time quantitative polymerase chain reaction
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Shanghai, China), and cDNA was synthesized using SMARTer PCR cDNA Synthesis Kit (Takara, Beijing, China) according to protocols recommended by the manufacturer. Polymerase chain reaction (PCR) primers used for different targets were listed in S2 Table. Subsequent quantitative reverse transcription PCR (RT-qPCR) was performed using QuantiFast one-step SYBR Green RT-PCR kit in Applied Biosystems 7300 Real-Time PCR System (Foster City, CA), as described in our previous work [14]. 18S served as the internal control.
3. Immunohistochemistry
Immunohistochemical staining was performed as previously described [15], with the aid of VECTASTAIN Elite ABC HRP Kit (Vector Laboratories, Burlingame, CA). The antibody used was rabbit anti-PITX2 polyclonal Ab (Abcam, Shanghai, China).
5. Cells treatment
HeLa cells and the ER-positive hormone-dependent MCF7 BCa cells were obtained from American type culture collection (ATCC, Manassas, VA). Cells were routinely cultured in Dulbecco's modified Eagle's medium medium supplemented with 10% fetal bovine serum (FBS; GIBCO, Shanghai, China) and 1% penicillin/streptomycin in a 37°C, 5% CO2 incubator. The generation of letrozole-resistant MCF7/LR cells has been described in our previous work [17]. MCF7/LR cells were maintained in phenol red-free improved minimal essential medium supplemented with 5% charcoal/dextran-treated FBS, 1% penicillin/streptomycin, 100 μg/mL hygromycin (Thermo Fisher Scientific, Shanghai, China), and 1 μmol/L of letrozole (Sigma-Aldrich, Shanghai, China). To overexpress the exogenous PITX2, MCF7 cells were transfected with pPM-His-PITX2 or pPM-His vector (GenScript, Nanjing, China) using Lipofectamine 3000 (Thermo Fisher Scientific), followed by Neomycin selection (200 μg/mL, Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. To stably knockdown the endogenous expression of PITX2, MCF7/LR cells were transfected with PITX2 shRNA or scramble shRNA (SABioscience, Shanghai, China) using Lipofectamine 3000. One day after transfection, the transfected cells were selected with 1.0 μg/mL puromycin (Sigma-Aldrich) for 1-2 weeks. To transiently knockdown the expression of IRF-7 or IFITM1, MCF7/LR and MCF7/LR/His-PITX2 cells were transfected with IRF-7 siRNA/Ctrl siRNA and IFITM1 siRNA/Ctrl siRNA (Santa Cruz Biotechnology, Shanghai, China), respectively. Forty-eight hours after transient transfection, cells were harvested for further analysis. To determine the potential regulation of PITX2 expression by IFNα signaling, BCa cells were challenged for 24 hours with a gradual concentration of recombinant hIFNα Protein (R&D Systems, Minneapolis, MN), in the presence or absence of the pretreatment with 5 μg/mL of anti-IFNAR neutralizing antibody (Millipore, Temecula, CA) for 4 hours.
6. Cell viability and apoptosis
BCa cells with different transfections were plated onto a 96-well plate at of density of 1×104 cells/well. After a 3-day culture and a following overnight starvation, cells were treated with 10–5 M of letrozole, along with 25 nM of androstenedione (Δ4A, Sigma-Aldrich), for another 4 days. Viable cell numbers were determined using trypan blue staining (Invitrogen) along with hemocytometer count (Beckman Coulter, Fullerton, CA) and cell apoptosis were assayed using an apoptosis enzyme-linked immunosorbent assay (ELISA) kit (Roche Diagnostics, Shanghai, China) [17,18], as per the manufacturer’s instructions.
7. In vivo letrozole sensitivity assay
Tumor growth in nude mice was performed according to our previous work [14]. Briefly, BCa cells with different transfections were resuspended in Matrigel (10 mg/mL, BD Biosciences, Shanghai, China). One hundred microliters of cell suspension (2×107 cells/mL) were then injected into left flanks of female nude mice. Four weeks after cells inoculation, mice were injected subcutaneously with vehicle, 100 μg/day of Δ4A, orΔ4A plus 10 μg/day of letrozole on a daily basis, for 32 days (Δ4A is the aromatase substrate and athymic mice are deficient in adrenal androgens [2]). Tumor volume changes were recorded every 3 days from the 14th day by two perpendicular diameter measurements.
8. Cell invasion assay
BCa cells (1×104) were cultured in the upper chamber of Transwell with a membrane coated with Matrigel for 24 hours, followed by treatment with 10–5 M of letrozole, along with 25 nM of Δ4A, or with dimethyl sulfoxide (Sigma-Aldrich) for 48 hours. Subsequently, the upper chamber were carefully removed and the remaining cells on the membrane were stained for 10 minutes with 0.1% crystal violet solution (Sigma-Aldrich). Final spectrophotometry was developed at 570 nM.
9. Measurement of human IFNα content
BCa cells were seeded in six-well plates and cultured overnight, followed by stimulation with 250 U/mL of recombinant hIFNα for 24 hours. Cell lysates and culture supernatants were then harvested and subjected to ELISA analysis using a commercial kit (Thunderscience, Shanghai, China). The lower limit of detection for hIFNα is 1.04 pg/mL, with a standard curve range of 15.63-1,000 pg/mL.
10. Immunofluorescence staining
BCa cells grown on coverslips were fixed with 4% formaldehyde diluted in phosphate buffered saline (PBS) at room temperature (RT) for 15 minutes. After a thorough rinse, cells were incubated with blocking buffer containing 1× PBS, 5% normal donkey serum and 0.3% Triton X-100 (Vector Lab, Burlingame, CA) at RT for 60 minutes, followed by incubation with the primary antibodies (S3 Table) at 4°C overnight. On the second day, cells were incubated with fluorochrome-conjugated second antibody for 1-2 hours at RT in the dark. Final immunofluorescence was observed under an inverted microscope (Axio Imager M1 microscope, Zeiss, Gottingen, Germany).
11. Promoter reporter assay
For the assessment of IFITM1 promoter activity, the genomic fragments harboring the putative PITX2-binding sites in human IFITM1 promoter (~1.8 kb upstream of the translation start site), were subcloned into the pGL4-Luc reporter vector (Promega, Madison, WI) using Infusion 2.0 Dry-Down PCR cloning kit (Clontech, Shanghai, China). Promoter activity was further validated by mutation of the putative PITX2-binding site on the promoter at –406/–402 by replacing AATCC with AcgaC using the QuikChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA). For reporter assay, 0.5 μg reporter plasmid and pRL-TK Renilla reporter plasmid were co-transfected with pPM-His-PITX2 or pPMHis vector into HeLa cells using FUGENE (Promega). Forty-eight hours later, cells were treated for 4 hours with 20 U/mL of hIFNα, followed by luciferase activity measurements using a dual luciferase reporter assay kit (Promega).
12. Chromatin immunoprecipitation
MCF7/His-PITX2 cells were challenged for 4 hours with 20 U/mL of hIFNα [4], and cells were then crosslinked with 1% formaldehyde and sonicated. Soluble chromatin was immunoprecipitated and subjected to chromatin immunoprecipitation (ChIP), as described in detail in our previous work [14]. Primers used for ChIP and double ChIP were listed in S2 Table.
13. Statistical analysis
Statistical analyses were performed using SPSS ver. 15.0 (SPSS Inc., Chicago, IL). Statistical comparisons and analyses were performed by either Student’s t test or one-way ANOVA, followed by Tukey post-hoc analyses wherever appropriate. The relative correlation rate was determined using the Pearson chi-square test. Results are presented as mean±standard error of mean. and p < 0.05 was considered statistically significant.
14. Ethical statement
All procedures involving animals, strictly conformed to the Guide for the Care and Use of Laboratory Animals, were approved by the Animal Care and Use Committee (ACUC) of Cancer Hospital of China Medical University (CMU-2016-332-1).
The study protocol, strictly following the ethical standards of Helsinki Declaration, was approved by the Ethics Committee of Cancer Hospital of China Medical University. Written informed consents were obtained from all participants.
Results
1. PITX2 upregulation is associated with the pathogenesis of letrozole resistance
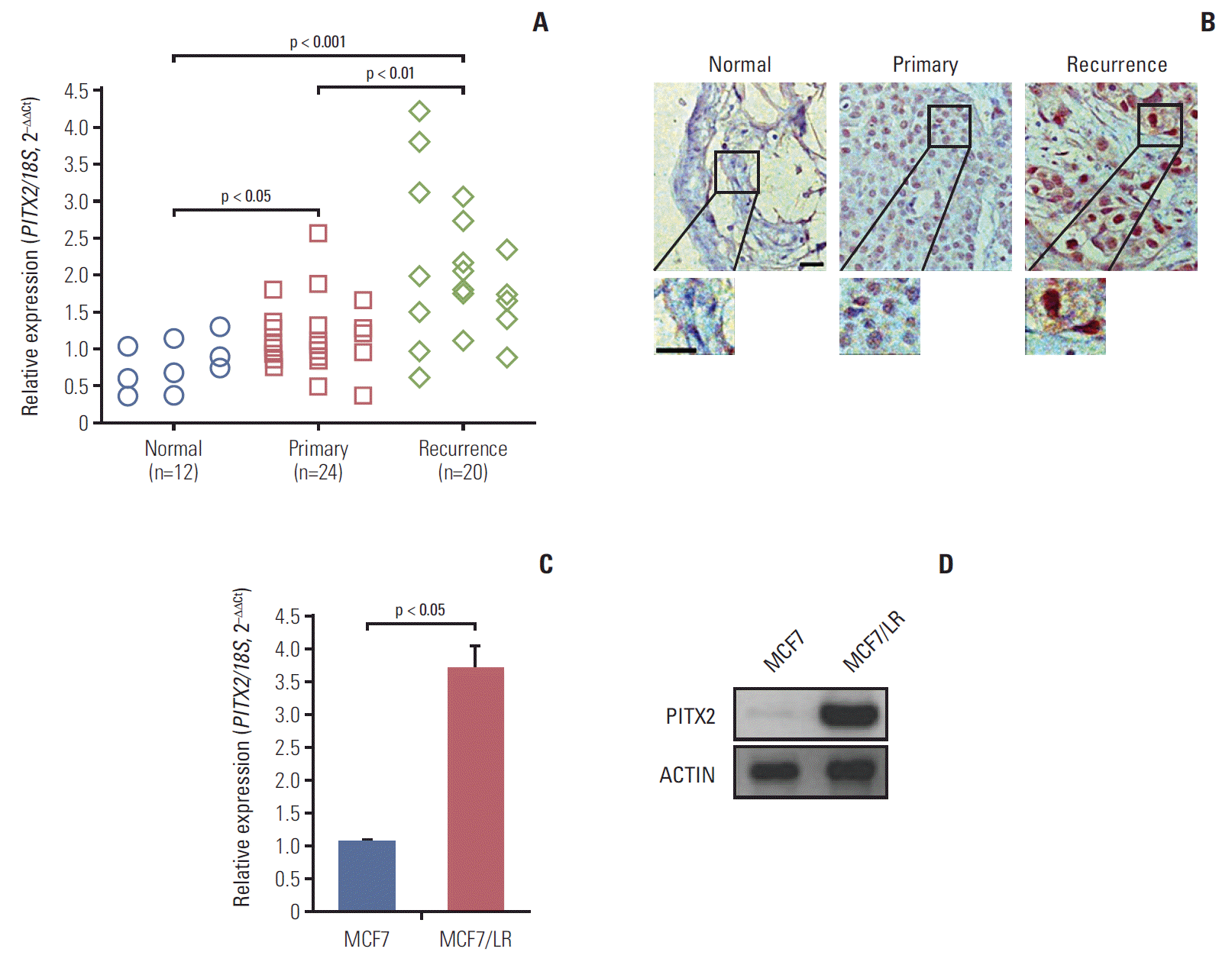
To examine the expression of PITX2 in human BCa tissues, RT-qPCR and immunohistochemical analyses were done on 12 cases of normal breast tissues, 24 cases of primary BCa biopses and 20 cases of letrozole-resistant (recurrent) BCa biopses. As shown in Fig. 1A, expression of PITX2 mRNA was significantly induced in recurrent BCa compared to other tissues. Histologically, PITX2 immunoreactivity, which was predominantly localized in the nuclear, was notably increased in recurrent BCa tissues (Fig. 1B). To further provide the in vitro evidence for the involvement of PITX2 in letrozole resistance, we examined the expression levels of PITX2 in letrozole-sensitive MCF7 cells and letrozole-resistant MCF7/LR cells. As expected, when MCF7 became letrozole-resistant, MCF7/LR cells showed an increased level of PITX2 expression (Fig. 1C and D). Thus, PITX2 upregulation correlates to the pathogenesis of letrozole-resistance in BCa.
2. Ectopic overexpression of exogenous PITX2 potentiates letrozole-resistance in MCF7 cells
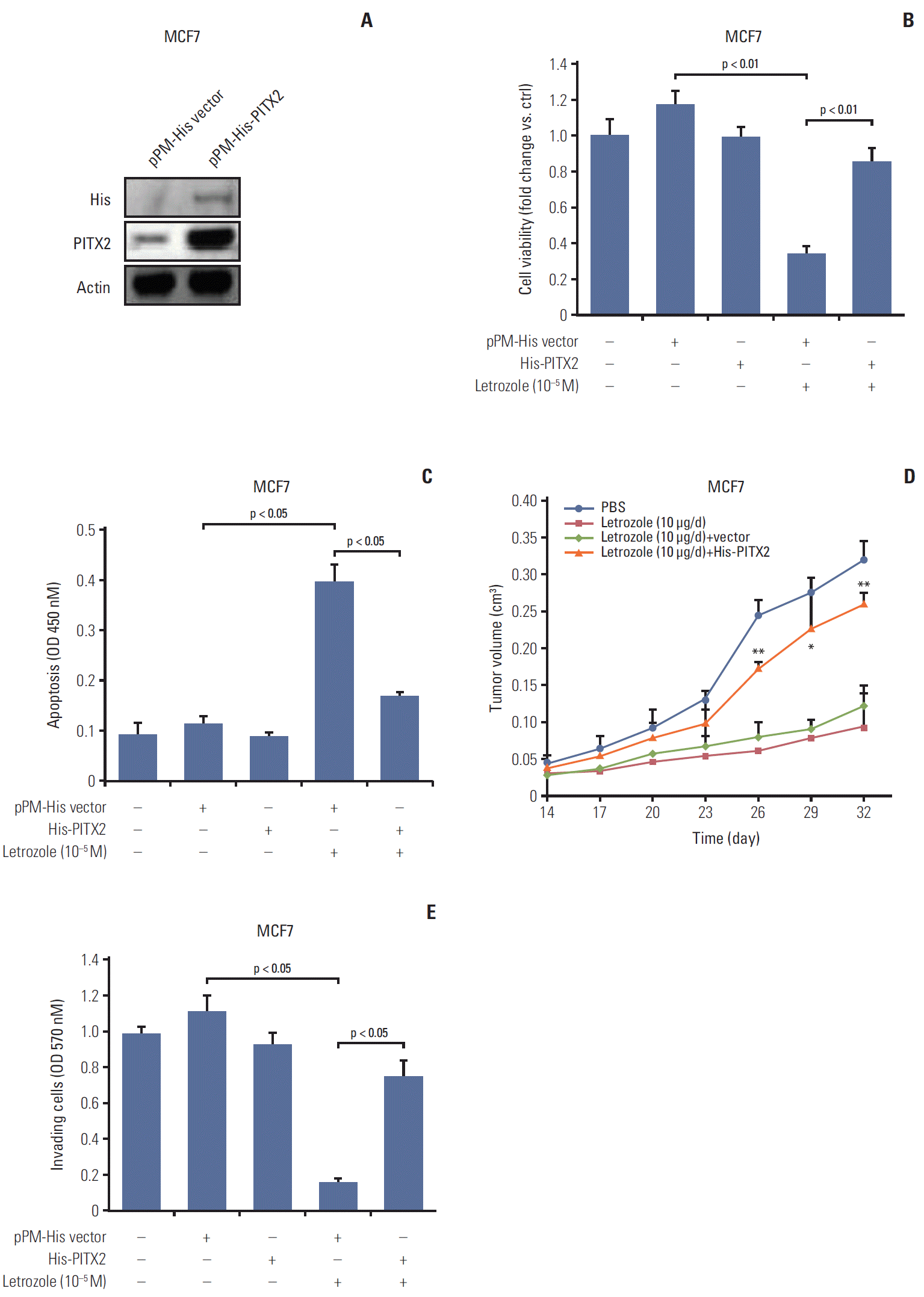
To directly ask whether PITX2 contributes to letrozole-resistance, we established MCF7 cells stably overexpressing pPM-His-PITX2 (Fig. 2A). The MCF7/His-PITX2 cells, along with the control cells, were then cultured in the medium containing 10–5 M letrozole. Letrozole-induced growth inhibition was totally reversed in MCF7/His-PITX2 cells, compared with the pPM-His vector–transfected cells (Fig. 2B). Of note, transfection with pPM-His-PITX2 or vector alone exerted no effects on cell viability. To investigate a PITX2-dependent regulation of anchorage-dependent tumor proliferation and cluster formation, we cultured MCF7/His-PITX2 cells, along with the control cells, in the medium containing basement membrane extract and 10–5 M letrozole for 16 days. The average size of tumor clusters with stable ectopic expression of His-PITX2 was significantly increased by ~1.8 fold as compared to His-vector-expressing MCF7 cells (S4A and S4B Fig.). Consistently, higher colony formation efficiency was observed after letrozole treatment in MCF7 cells overexpressing His-PITX2 when compared with His-vector-expressing MCF7 cells (S4C Fig.), suggesting that ectopic expression of exogenous PIXT2 can enhance anchorage-dependent and -independent growth of MCF7 cells upon letrozole challenge. On the other hand, letrozole-induced apoptosis was partially but effectively attenuated in MCF7/His-PITX2 cells (Fig. 2C). In agreement, the formation of xenograft tumors in letrozole-challenged immunodeficient mice were substantially augmented in MCF7 cells by PITX2 overexpression (Fig. 2D). Moreover, letrozole-elicited inhibition of cell invasiveness was completely abolished by PITX2 overexpression (Fig. 2E). Taken together, these data demonstrate that PITX2 directly promotes letrozole-resistance in ER-positive BCa cells.
3. Endogenous PITX2 protects MCF7/LR cells from letrozole toxicity
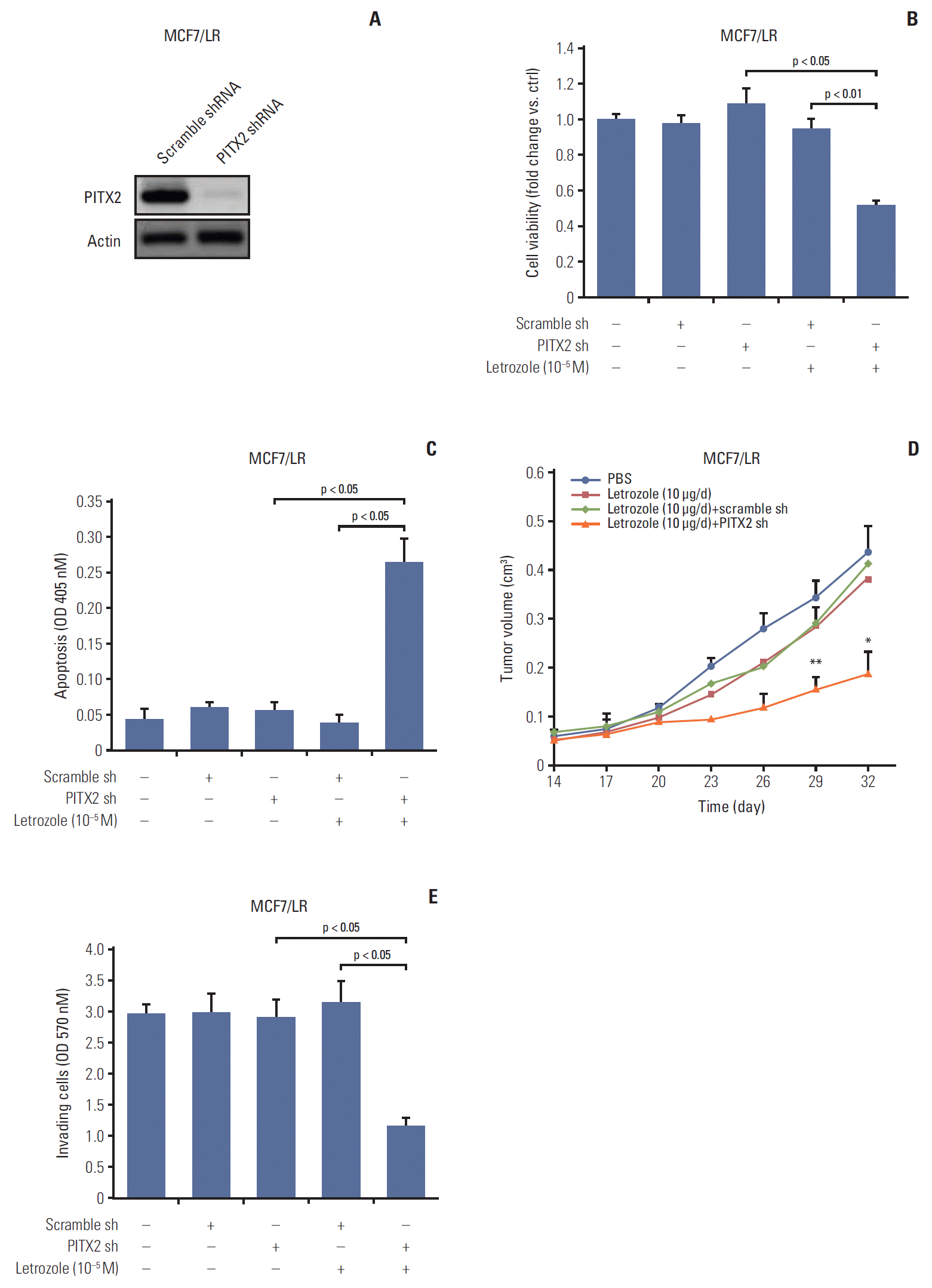
Having established the causal link between PITX2 expression and letrozole-resistance, we then determined the effects of PITX2 inhibition on the malignant status of MCF7/LR cells. The stable knockdown of PITX2 expression by shRNA was validated using immunoblotting (Fig. 3A). The letrozole-induced growth inhibition was significantly enhanced in the MCF7 cells depleted of endogenous PITX2, compared to that in cells transfected with Ctrl shRNA (Fig. 3B). Transfection with PITX2 shRNA or Ctrl shRNA alone exerted no effects on cell viability. We also confirmed the promoting effects of PITX2 on anchorage-dependent and -independent growth of MCF7/LR cells upon letrozole challenge, using tumor cluster formation assay (S4D and S4E Fig.) and soft agar colony formation experiments (S4F Fig.). Moreover, as shown by the apoptotic ELISA assay, upon letrozole challenge, MCF7/LR/PITX2 shRNA cells exhibited higher apoptosis (~6.8 fold) when compared with MCF7/LR cells transfected with scramble shRNA (Fig. 3C). Consistently, the formation of xenograft tumors in letrozole-challenged immunodeficient mice were significantly compromised in MCF7/LR cells by PITX2 ablation (Fig. 3D). Additionally, the cell invasiveness of MCF7/LR/PITX2 shRNA cells was significantly reduced by ~62.4% as compared to MCF7/LR cells transfected with scramble shRNA (Fig. 3E). Thus, repression of PITX2 expression may augment the inhibitory effect of letrozole on the proliferation and clonogenicty of BCa cells.
4. Intracellular IFNα signaling regulates PITX2 expression
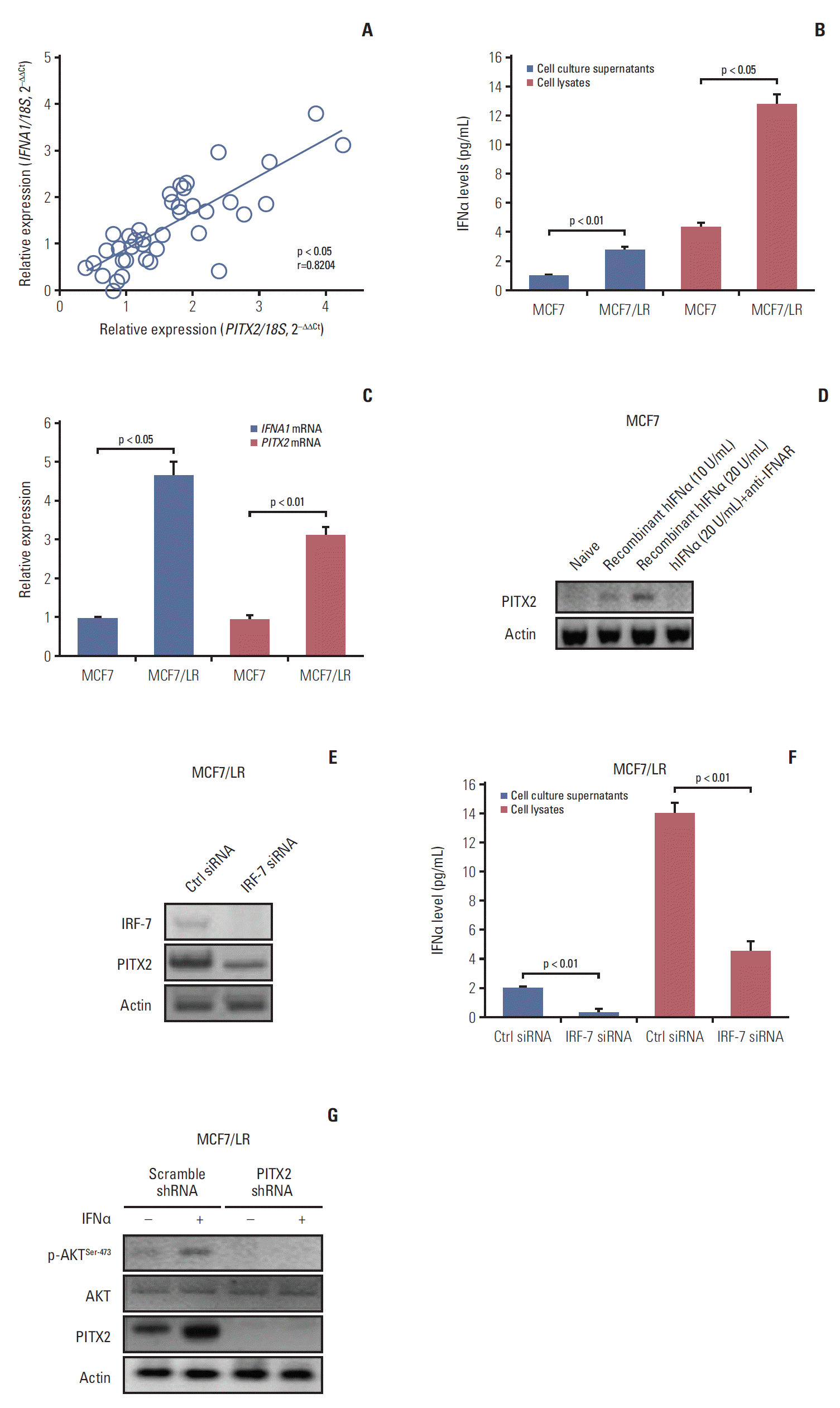
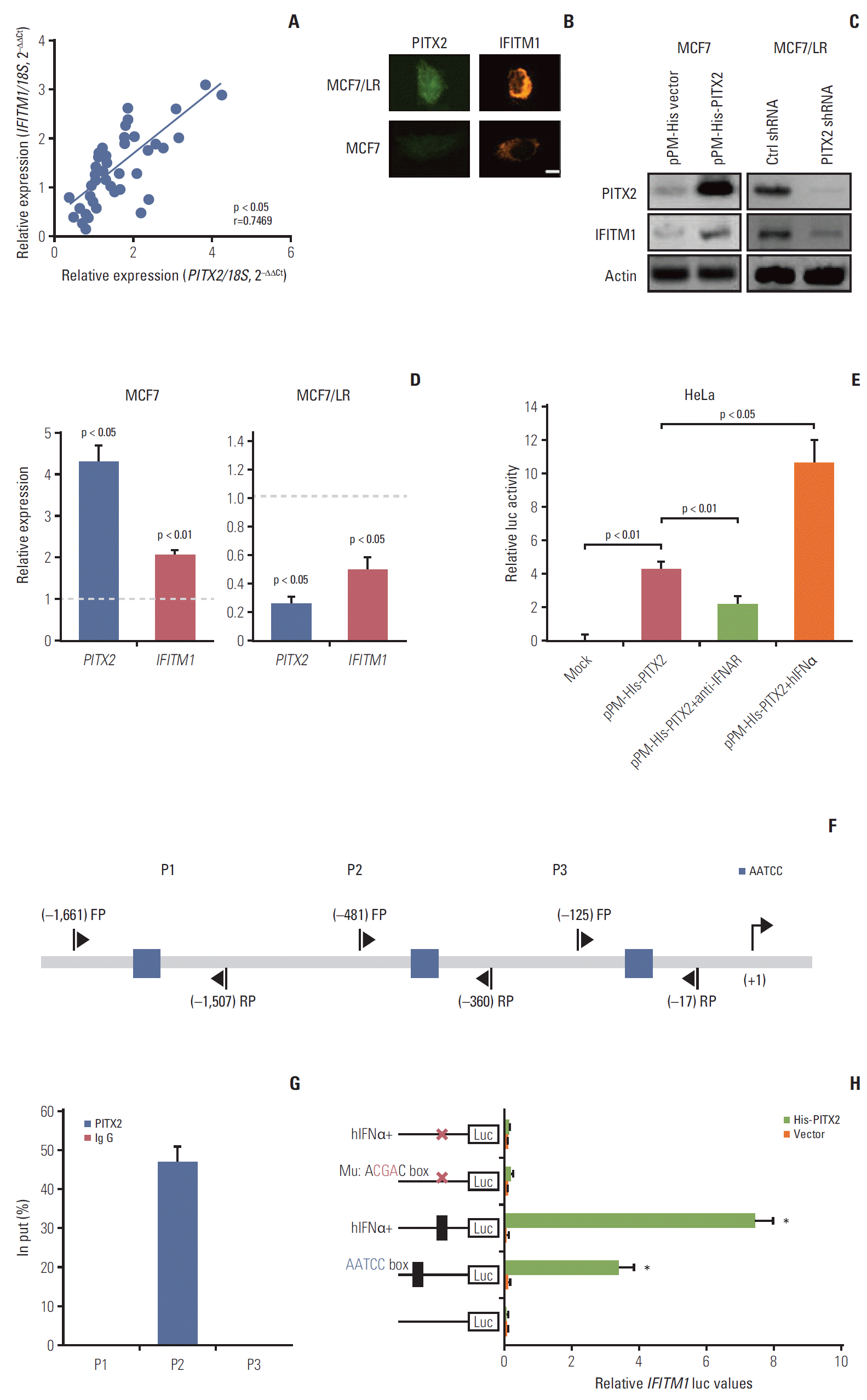
During the course of the study, we unexpectedly observed that PITX2 mRNA expression was positively correlated to IFNA1 (the gene encoding IFNα) mRNA level in the abovementioned 44 cases of BCa samples (Fig. 4A). Because the IFNα signaling pathway plays a pivotal role in the regulation of tumorigenesis and drug resistance, understanding how IFNα regulates the cellular processes controlling malignant transformation holds a lot of promise in combating BCa [4]. To this end, we then determined the expression levels of IFNα protein in different BCa cells using a ELISA kit. As shown in Fig. 4B, IFNα content was significantly higher in the supernatant and lysates of MCF7/LR cells than that in MCF7 cells. Consistently, MCF7/LR cells exhibited higher expression levels of both PITX2 mRNA and IFNA1 mRNA, when compared to the parental MCF7 cells (Fig. 4C). To directly assay the regulation of PITX2 expression by IFNα signaling, we treated MCF7 cells with gradual doses of recombinant hIFNα. IFNα induced PITX2 expression in a dose-dependent manner. In contrast, blocking IFNα receptor using a previously-validated neutralizing antibody effectively abolished IFNα-elicited PITX2 induction [4] (Fig. 4D). Considering that endogenous IFNα production is predominantly controlled at the transcriptional level by IRF-7 [4], we then transiently knocked down the IRF-7 expression using siRNA in MCF7/LR cells (Fig. 4E). Interestingly, ablation of IRF-7 significantly reduced endogenous IFNα production in both cell supernatant and lysates (Fig. 4F). Of particular interest, PITX2 repression substantially eliminated the IFNα-stimulated phosphorylation of AKTSer-473 (Fig. 4G). Thus, PITX2 and IFNα signaling pathways may interact with each other in a reciprocal manner.
5. PITX2 transcriptionally regulates IFITM1 expression
In search of the specific component of IFNα pathway that may underlie PITX2 function, we observed that the mRNA expression of IFITM1 (a well-known IFN response gene) was positively correlated to PITX2 mRNA level in the above-mentioned 44 cases of BCa samples (Fig. 5A). In agreement, a strong nuclear staining of PITX2 protein was associated with a clear cytoplasm and membrane staining of IFITM1 protein in MCF7/LR cells, whereas a negligible staining of both PITX2 and IFITM1 proteins was frequently observed in MCF7 cells (Fig. 5B). In MCF7 cells, overexpression of the exogenous PITX2 resulted in a significant induction of IFITM1 expression at both transcriptional and translational levels, whereas knockdown of PITX2 by shRNA significantly compromised PITX2 and IFITM1 expression levels (Fig. 5C and D). To study whether PITX2 directly regulates IFITM1 transcription, we performed luciferase reporter assays in HeLa cells using the fragments harboring the putative PITX2-binding sites (AATCC) in IFITM1 promoter. Transfection with pPM-His-PITX2 significantly increased the IFITM1 reporter activity relative to control vector. This stimulatory effect was further amplified by co-incubation with hIFNα. In good contrast, blocking IFNα receptor using a previouslyvalidated neutralizing antibody effectively abolished the pPM-His-PITX2–induced IFITM1 reporter activity, indicating that IFNα signaling is involved in the transcriptional activation of IFITM1 by PITX2 (Fig. 5E). Notably, analysis of the 5′ flanking regions of human IFITM1 identified three sequences containing the conserved binding site of PITX2 (AATCC box) (Fig. 5F). We then tested whether PITX2 functions as a direct regulator of the putative binding sites in the IFITM1 promoter by using ChIP assay. PITX2 could bind the promoter region that harbors the proximal AATCC box (–645/–641) (Fig. 5G). Furthermore, mutation of the binding site by replacing AATCC with AcgaC using the site-directed mutagenesis greatly compromised PITX2-enhanced promoter activity, even in the presence of hIFNα stimulation (Fig. 5H). Additionally, to clarify the regulatory relationship between IFN and IFITM1 upon PITX2 overexpression at the in vivo level, we subjected the MCF7/His-PITX2 cells, along with the His-vector-expressing MCF7 cells to in vivo letrozole sensitivity assay. As expected, PITX2 overexpression caused a significant increase in both IFNα and IFITM1 expression levels. The induction in the IFITM1 expression levels in xenograft tissues was further augmented by letrozole treatment. In contrast, treatment with 10 μg/day of letrozole for 2 weeks could not stimulate futher the IFNα production in xenograft tissues (S5 Fig.). Thus, PITX2 positively regulates IFITM1 transcription by direct binding its chromatin.
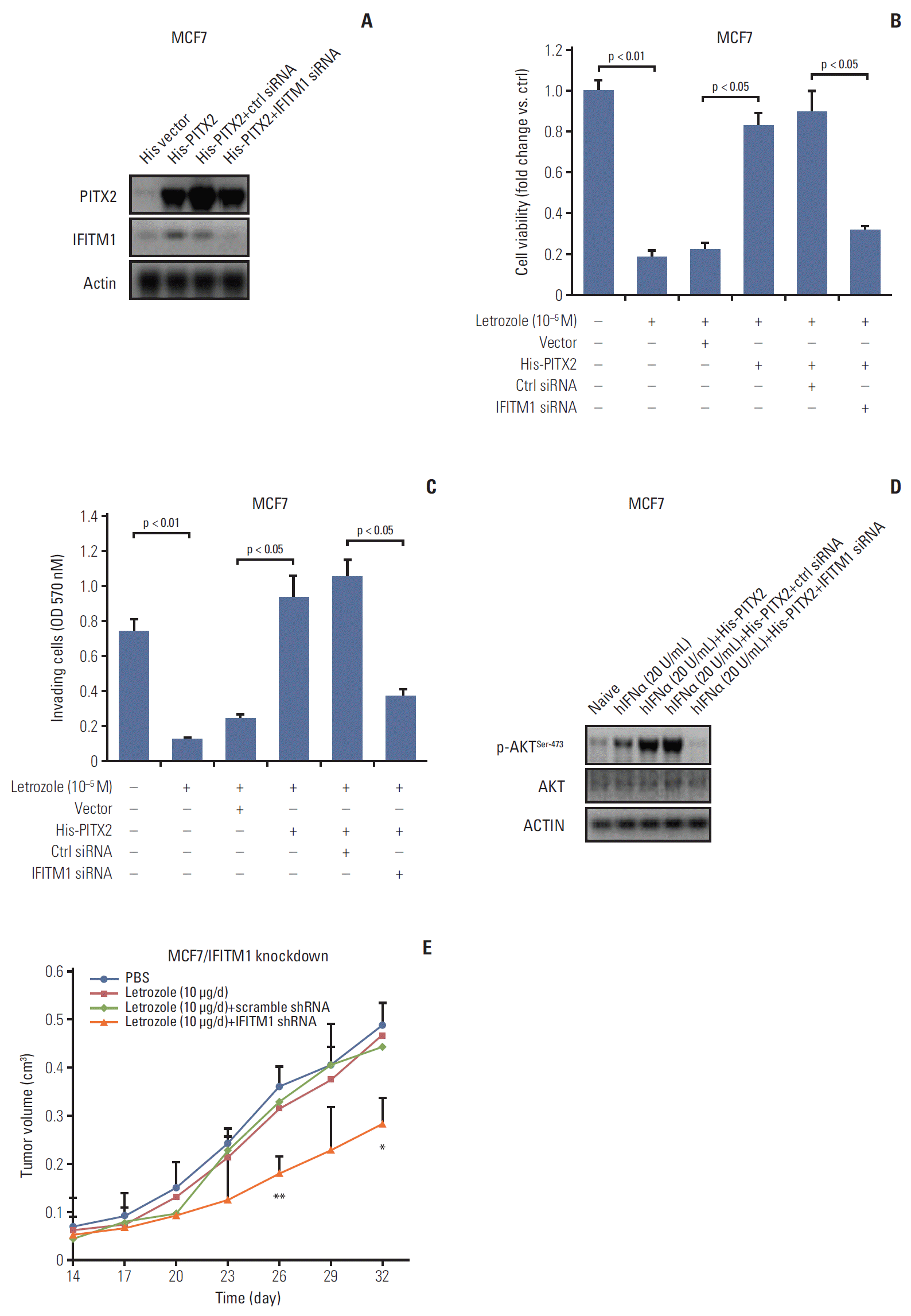
6. Repression of IFITM1 expression abolished PITX2-induced letrozole resistance
In the last experimental setting, we determined whether IFITM1 alone was sufficient to explain PITX2-induced letrozole resistance. Transient transfection in MCF7/His-PITX2 cells with IFITM1 siRNA significantly decreased IFITM1 expression, but exerted no effects on PITX2 expression levels (Fig. 6A), reemphasizing the notion that PITX2 acts upstream of IFITM1 signaling pathway. Incubation with 10–5 M letrozole for 4 days resulted in a remarkable decrease of cell viability, whereas stable expression of PITX2 caused an obvious letrozole resistance by enhancing viability. More importantly, transient knockdown of IFITM1 expression could effectively abolish PITX2 overexpression-induced impact on cell viability, and partially recovered MCF7/His-PITX2 cells sensitivity to letrozole (Fig. 6B). To determine whether these in vitro associations between letrozole resistance and disruption of PITX2/IFITM1 pathway also affect cell invasiveness, we performed Transwell cell invasion assay. Ectopic expression of exogenous PITX2 significantly enhanced in vitro cell invasiveness upon letrozole challenge, and this letrozole resistance was effectively but partially reversed by transient knockdown of IFITM1 expression using siRNA (Fig. 6C). In agreement, ablation of IFITM1 expression substantially abolished PITX2 overexpression-elicited phosphorylation of AKTSer-473 (Fig. 6D). Lastly, by using the MCF7/LR cells in a xenograft model, we verified that ablation of endogenous IFITM1 expression could successfully but partially restore the responsiveness to letrozole treatment in MCF7/LR cells (Fig. 6E, S6 Fig.). Taken together, the available data confirm the relevance of the disruption of the PITX2/IFITM1 pathway in conferring letrozole resistance.
Discussion
The homeodomain transcription factor PITX2 was originally identified as an indispensible regulator for extraocular muscle development. However, emerging data suggest that additional, as yet poorly understood, non-muscle actions of PITX2 are possible to take place. For example, deregulation of PITX2 expression has been frequently reported in bladder cancer [19], esophageal squamous cell carcinoma [20], renal cancer [11], endometrioid endometrial adenocarcinoma [21], prostate cancer [22], and BCa [23]. In the present study, evidence is provided that PITX2 upregulation promotes the pathogenesis of letrozole resistance in BCa. We also confirmed the oncogenic effects of PITX2 signaling in another two ER-positive BCa cells (namely MDA-MB-415 and MDA-MB-415/LR) (Figs. 4-6, S7-S9 Figs.). Thus, PITX2 may serve as an important regulator of both muscle development and transformed cancer cells.
PITX2 expression responding to different cellular context is rigorously regulated by epigenetic mechanisms. For example, because PITX2 methylation significantly correlates to clinicopathologic parameters, hypermethylation of this homeodomain transcription factor has been suggested to be a strong predictor for recurrent prostate cancer. In agreement, hypermethylation of PITX2 promoter has been observed to be closely associated with BCa grade and stage [24]. These two observations are strongly indicative of that PITX2 expression should be much lower in the highly malignant cancer cells. In our study, however, PITX2 expression was substantially upregulated in letrozole-resistant BCa tissues (Fig. 1A and B). Despite different methodologies employed, three possibilities may account for this discrepancy. First, BCa is a heterogeneous disease entity. The high degree of diversity between and within BCa cells differentiates the risk of disease progression and therapeutic resistance [25]. Considering that letrozole-resistant BCa cells exhibit differential cellular behaviors and genome characteristics, it is very likely that PITX2 may exhibit a dual function in response to distinct cell contexts depending upon the state of cell malignancy. Second, hypermethylation of a promoter does not necessarily repress the gene expression. Thyroid-stimulating-hormone α (TSH-α) is such a striking example. Two CpG sites in the TSH-α promoter is hypermethylated in thyroid cancer cells but the expression of TSH-α in thyroid cancer remains relatively high when compared to that in normal thyroid tissues [26]. Lastly, besides promoter hypermethylation, other epigenetic mechanism(s) may coexist to determine the level of PITX2 expression. For instance, using miRNA profiling and pharmacological assay, Zhang et al. [27] have identified miR-644a as a negative mediator of PITX2 during the pathogenesis of esophageal squamous cell carcinoma. Similarly, in cardiomyocyte background, miR-21 upregulation may directly target PITX2, thereby underlying the onset of atrial fibrillation [28]. Thus, the duality of PITX2 function in transformed cells may be considered as an extension of the complexity of different co-regulators.
Evidence from both experimental and clinical studies have confirmed that activation of the PI3K/AKT pathway, functioning as an indispensible adaptive mechanism, is a hallmark of resistance to cancer treatment. Specifically in BCa, treatment with AKT siRNA or with the PI3K inhibitor taselisib both effectively impair the cell viability of letrozoleresistant cells, thus overcoming acquired letrozole resistance [29]. Interestingly, there is increasing evidence that intensive crosstalk between IFNα/γ and AKT signaling is involved in cancerous progression. Activation of the AKT pathway by the IFNα/γ complements the function of IFN-activated downstream pathways, and ultimately mediates the biological effects of IFN signal [30]. However, how the AKT pathway is delicately coordinated in the induction of IFN-mediated biological responses, especially under certain pathophysiological conditions, remain largely unknown. The data shown here reveal that PITX2 is required for the IFNα-stimulated activation of the AKT pathway in letrozole-resistant BCa cells (Fig. 4G). It will be of future interest to deconvolute whether PITX2 alone is sufficient enough to explain the IFNα-induced phosphorylation of AKT during the pathogenesis of drug resistance.
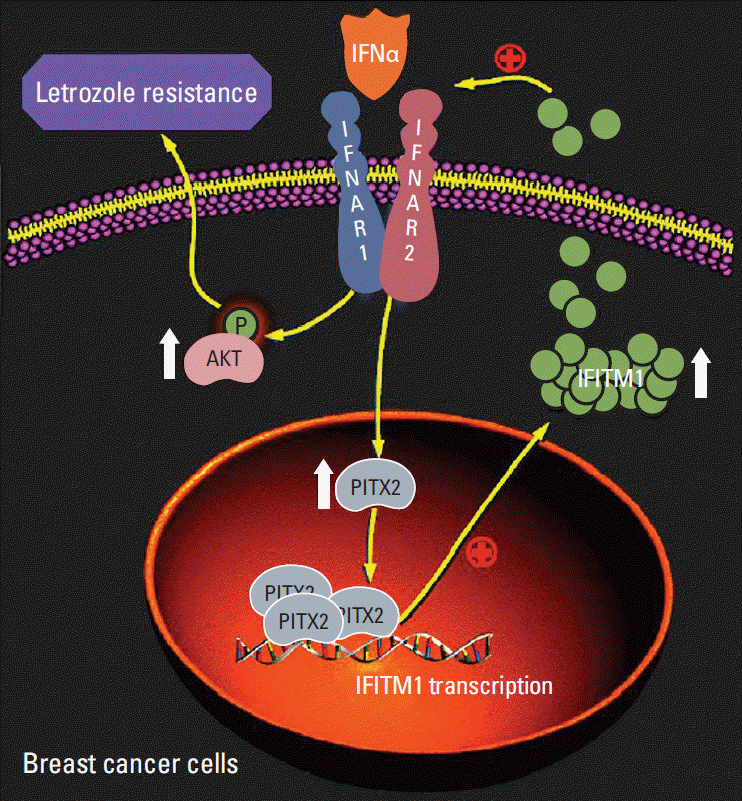
Another relevant central question addressed by the current study is how PITX2 regulates IFNα-elicited cellular response. We have shown that PITX2 activated IFITM1 transcription by directly binding its promoter (Fig. 5E-G), and this chromatin recruitment of PITX2 was significantly augmented upon hIFNα stimulation (Fig. 5H). More importantly, ablation of IFITM1 could effectively abolish PITX2 overexpression-induced letrozole-resistance (Fig. 6B and C). Thus, PITX2 may serve as a potent positive regulator of letrozole-resistance in BCa cells, likely functioning at least in part via transactivation of IFITM1 signaling. IFITM1 is a transmembrane protein that is strongly induced by IFNα and IFNβ. Recent advance in this field have revealed that IFITM1, in turn, could enhance IFNα-mediated biological responses in AI resistant BCa cells [4]. In favor of the latter point, we observed that knockdown of IFITM1 dramatically reduced IFNα-elicited phosphorylation of AKT, even in the presence of PITX2 overexpression (Fig. 6D). These findings together reveal a critical homeostatic role of PITX2/IFITM1, both as a target and as a component of the IFNα circuitry, in the regulation of inflammatory responses of letrozole-resistant BCa cells, likely affecting the activation of the AKT pathway (Fig. 7). Of particular interest, a putative cis-regulatory element has been previously identified in the IFITM1 promoter region (–37/+181 bp) [31]. The signal transducers and activators of transcription 1 (STAT1) binds to this region and is required for IFITM1 transactivation. In contrast, the promoter region of IFITM1 bound by PITX2 (–406/–402 bp) was not included in the above-mentioned cis-acting element. Therefore, our results help to broaden the understanding of the diversity of the transcriptional regulation sites of IFITM1 expression.
In summary, the data presented here have demonstrated that induction of PITX2 expression is an intrinsic molecular event during the pathogenesis of letrozole-resistance. As a mechanism of direct transactivation of IFITM1 by PITX2, the PITX2/IFITM1 cascade, both as a target and as a modifier of the IFNα pathway, in conferring an adaptive mechanism upon therapeutic stress. As a result, PITX2 enhances cell survival, augments AKT activation and potentiates cell invasiveness in the presence of letrozole treatment, all of which are characteristic features of letrozole-resistant BCa. Thus, our findings of PITX2 regulation of IFITM1 expression introduce a new regulatory player of IFNα pathway and in principle identify PITX2/IFITM1 cascade as a potential new therapeutic target for letrozole-resistant BCa.
 XML Download
XML Download