

 PDF
PDF Citation
Citation Print
Print


Introduction
Intense Helicobacter pylori infection promotes a continuum of various macroscopic and histological changes in the gastric mucosa, including severe atrophic gastritis, intestinal metaplasia, dysplasia, and gastric cancer [1-4]. Intestinal metaplasia and dysplasia are considered premalignant lesions [2]. However, approximately half of gastric cancer patients do not show intestinal metaplasia or dysplasia in background gastric mucosa [5]. Even adjacent pairs of dysplasia and cancer tissues fail to display same mutations [6]. This raises a question over whether H. pylori infection induces dysplastic and neoplastic transformation of epithelial cells individually or sequentially. Intense H. pylori infection that can elevate the risk of gastric cancer tends to disappear when progression of gastric mucosal atrophy with age creates poor tissue environment for bacterial colonization [1,3]. Therefore, a surrogate marker is required to identify an individual who has previously experienced H. pylori infection with cancer risk.
Glandular stem cells maintaining gastrointestinal mucosa are periodically replaced with new stem cells. For example, an old glandular stem cell in each colonic gland is replaced by a new one every eight years [7]. Pluripotent mesenchymal cells derived from various organ sites including adipose and bone marrow tissues can become new stem cells when native gastric stem cells are damaged by inflammation or when they lose their regenerative capacity [8]. Newly adapted stem cells are capable of restoring undifferentiated histology and may be transformed into gastric cancer stem cells. Because mice rarely develop invasive gastric cancer [9], few mouse models have confirmed the neoplastic transformation of marrow-derived stem cells [10]. Humans and mice have distinct epigenetic structures around housekeeping genes. Epigenetic traits of housekeeping genes in humans can promote stem cell replacement but predispose stem cells to reactivating intrinsic migration traits [11]. Undifferentiated-type gastric cancer is common in relatively younger individuals, compared to differentiated-type cancer [12]. This suggests that new stem cells at an early stage of adaptation can preserve marrow properties. They are prone to transform into diffuse-type gastric cancer. Meanwhile, new stem cells at a late stage of adaptation are able to trans-differentiate into intestinal-type gastric cancer when they acquire gastric stem cell properties [13]. It is likely that the development of gastric cancer is associated with phenotypic stability of periodically replaced stem cells.
In H. pylori-infected stomach, numerous housekeeping genes containing CpG islands are concurrently methylated in an age-related manner [14,15]. It has been thought that concurrent CpG-island methylation can stabilize newly established phenotypes of stem cells [11,16,17]. Such agerelated changes in CpG-island methylation will reach a peak during new stem cell replacement [14]. Gastric mucosal atrophy spreading from the antrum to the body appears to impede phenotype-stabilizing methylation largely in the antrum [15]. This can result in insufficient methylation in the cancer-prone antrum [15,18], thus raising the epigenetic risk of gastric cancer in association with mucosal atrophy. Meanwhile, the gastric body with a minimal mucosal atrophic effect exhibits frequent CpG-island overmethylation in patients with cancer, indicating that a large number of methylation-inducible new stem cells are present over the background mucosa [15]. Therefore, cancer-related period of stem cell replacement can be estimated by analyzing the period of CpG-island overmethylation and the level of gastric mucosal atrophy.
There are transitional-CpG sites on methylation gradients between weakly methylated transcriptional start sites (TSS) and densely methylated retroelements [16,17,19,20]. These transitional-CpG sites display age-related methylation changes in the H. pylori–infected stomach, providing useful epigenetic information about the period of stem cell replacement [14,15]. The window period between H. pylori infection and cancer appearance, during which H. pylori infection disappeared with the progression of gastric mucosal atrophy, was investigated based on the age of transitional-CpG overmethylation.
Materials and Methods
1. Patients and biopsy specimens
A total of 205 healthy individuals, 99 patients with gastric cancer, and 118 patients with gastric dysplasia were enrolled in this study (Table 1). Controls and patients underwent endoscopic biopsy at St. Paul’s Hospital of The Catholic University of Korea between March 2008 and October 2013. Pairs of gastric biopsies were obtained from greater curvatures of the middle body and lesser curvatures of the antrum (1-2 cm distal from the angle). For patients with cancer or dysplasia, normal-appearing gastric mucosa located more than 2 cm from pathologic lesions was sampled to ensure epigenetic purity of normal mucosa cells [15]. H. pylori infection was examined using the Warthin-Starry silver impregnation method. The extent of gastric mucosal atrophy was evaluated using the endoscopic atrophic border score proposed by Kimura and Takemoto [21]. It was grouped into four levels of severity: none, closed-0 type; mild, closed-1 and closed-2 types; moderate, closed-3 type; and severe, open type (S1 Fig.). The level of gastric mucosal atrophy was measured repeatedly by Dr. Jung-Hwan Oh who is an experienced gastroenterologist.
Table 1.
Descriptive characteristics of study participants
| Control (n=205) | Cancer (n=99) | Dysplasia (n=118) | |
|---|---|---|---|
| Age (yr) | 54±10a) | 62±11 | 66±9a) |
| Sex | |||
| Male | 104 (51) | 79 (80) | 88 (75) |
| Female | 101 (49) | 20 (20) | 30 (25) |
| p-valueb) | < 0.001 | < 0.001 | |
| Helicobacter pylori | |||
| Negative | 95 (46) | 64 (65) | 76 (64) |
| Positive | 110 (54) | 35 (35) | 42 (36) |
| p-valueb) | 0.003 | 0.002 | |
| Intestinal metaplasia | |||
| No | 89 (43) | 20 (20) | 22 (19) |
| Yes | 116 (57) | 79 (80) | 96 (81) |
| p-valueb) | < 0.001 | < 0.001 | |
| Atrophic level | |||
| None | 16 (8) | 0 | 0 |
| Mild | 97 (47) | 27 (27) | 23 (19) |
| Moderate | 71 (35) | 31 (31) | 29 (25) |
| Severe | 21 (10) | 41 (42) | 66 (56) |
| p-valueb) | < 0.001 | < 0.001 |
![]()
2. DNA extraction and bisulfite treatment
DNA preparation and sodium bisulfite modification were performed as described elsewhere [15,22]. Biopsy specimens were dissolved in proteinase K lysis buffer and incubated at 37°C for 24 hours. Genomic DNA was extract from biopsy specimen using DNA isolation kit (A1120, Promega, Madison, WI) according to the manufacturer’s instructions. One microgram of DNA was denatured by treatment with 3 M NaOH followed by treatment with 2.3 M sodium bisulfite for 12 hours. DNA sample was purified using DNA purification resin (A1120, Promega), treated with 3 M NaOH, precipitated with ethanol, and resuspended in 40 μL of 5 mM Tris buffer. Finally, 1 μL aliquots were used as templates for polymerase chain reactions (PCRs).
3. Radioisotope-labeling methylation assay
Methylation-specific PCR (MSP) was performed in 10 μL of PCR reaction mixture containing 1 μL of bisulfite-modified DNA, 1 mCi of a-32P dTTP (PerkinElmer, Boston, MA), 62.5 mM of dATP, dCTP, and dGTP, 25 mM of dTTP, 1 pM of primers, 0.1% Tween 20 and 0.3 unit of Taq DNA polymerase (Takara, Kyoto, Japan). Thermal cycles were as follows: denaturation at 99°C for 5 minutes; addition of DNA polymerase and dNTP mixture; amplification for 32 cycles at 93°C for 50 seconds, 53-62°C for 1 minute, and 72°C for 1 minute; and extension at 72°C for 3 minutes. MSP products were subjected to polyacrylamide gel electrophoresis and visualized by repeated autoradiography using a radioluminograph scanner (PMI 170-9400, BioRAD, Hercules, CA). Band intensities were analyzed with TINA image software (Raytest Isotopenme-gerate, Straubenhardt, Germany). Methylation densities were calculated by dividing intensities of methylation primer sets by total intensities of unmethylation and methylation primer sets. The methylation status of each gene was classified into 10 levels with 10% methylation increments. Cut off point of gene overmethylation was determined on the basis of two intermediate methylation levels common in H. pylori–negative mucosa (S2 Table).
4. Methylation analysis of small biopsy specimens
A total of 16 methylation-variable transitional-CpG sites were chosen from eight housekeeping genes containing CpG islands (CDH1, ARRDC4, PPARG, TRAPPC2L, MMP2, CDKN2A, RUNX2, and RUNX3) and eight tissue-specific genes lacking CpG islands (TFF2, TFF3, TFF1, PGC, ATP4B, GHRL, PGA, and APC) (S2 Table) [15,22]. MSP primers for these CpG-island genes spanned CpG-island margins between –3.1 to –0.1 kb relative to the TSS. MSP primers for CpGisland-lacking genes were designed at –0.2 and 0 kb relative to the TSS.
Low GC content and repetitive sequences in the methylation-variable transitional-CpG sites can interfere with specific template-primer binding during PCR amplification. When using non-radioisotope PCR, MSP primers designed in the transitional-CpG sites are prone to generate non-specific amplicons [14]. Quantitative methylation analysis requires sufficient starting template DNA. The small amount of DNA obtained from small biopsy tissue samples was insufficient for analysis of 16 transitional-CpG sites using non-radioisotope PCR.
Pyrosequencing methylation analysis was found to have a limitation in analyzing the transitional-CpG sites (S3 Fig.). Due to a high T content of bisulfite-modified sequences (51%), we failed to design primers for common PCR for the ARRDC4 transitional-CpG sites. Pyrosequencing of MMP2 transitional-CpG sites (T content, 44%) failed to obtain reproducible methylation results. The CDH1 transitional-CpG sites containing a low T content (39%) demonstrated similar methylation values between the pyrosequencing and radioisotope MSP methods [15]. The radioisotope-labeled MSP ensured specific amplification at all 16 transitional-CpG sites (S4 Fig.).
5. Statistical analysis
Methylation values between radioisotope MSP and pyrosequencing were compared using Spearman’s correlation coefficients. One-way ANOVA with Tukey’s post-hoc test was employed to compare age differences among subject groups. Frequencies of clinicopathological features among non-cancer controls, cancer patients, and dysplasia patients were compared using chi-square test. Mean numbers of overmethylated genes among subject groups were compared by one-way ANOVA followed by Tukey’s post-hoc test. Differences in overmethylation frequencies between the antrum and body were analyzed using McNemar’s test. Age differences between two methylation groups were analyzed by Student’s t test. Multinomial logistic regression models were used to calculate odds ratios (ORs) and 95% confidence intervals (CIs) for the associations between LTR-dominant methylation type and cancer/dysplasia using controls as reference group. All ORs were adjusted by sex and age. p-values were two-sided. Statistically significant was considered at p < 0.05. All statistical analyses were conducted using SAS ver. 9.2 (SAS Inc., Cary, NC).
Results
1. Baseline characteristics
Baseline characteristics of study subjects (n=422) are summarized in Table 1. H. pylori infection was more (p < 0.01) frequently detected in control subjects than that in cancer patients or dysplasia patients. Control subjects (mean age, 54 years) were younger (p < 0.001) than cancer patients (62 years) and dysplasia patients (66 years). The level of gastric mucosal atrophy was higher (p < 0.001) in both cancer and dysplasia patients than that in controls. Severe atrophy was more frequent (p=0.033) in dysplasia patients than that in cancer patients.
2. Transitional-CpG methylation in the gastric antrum and body
The transitional-CpG sites of CpG-island genes were categorized into Alu-adjacent (CDH1, ARRDC4, PPARG, and TRAPPC2L) and LTR-adjacent (MMP2, CDKN2A, RUNX2, and RUNX3) genes based on the tendency for concurrent methylation depending on the type of gene-adjacent retroelements (S2 Table) [14,15]. Overmethylated genes in both cancer and dysplasia patients were counted regardless of H. pylori infection considering few differences in overmethylation frequencies between H. pylori–positive and H. pylori–negative patients (S5 Fig.). Mean numbers of overmethylated Alu- or LTR-adjacent genes were compared among two control groups (H. pylori–positive and –negative) and two patient groups (gastric cancer and dysplasia) (Fig. 1A). The number of overmethylated Alu-adjacent genes was high in the antrum of H. pylori–positive controls. LTR-adjacent overmethylation in the body was significantly (p < 0.001) increased in the two patient groups than that in H. pylori–positive controls. There was a significant (p < 0.001) decrease in Aluadjacent overmethylation in the antrum of the two patient groups. Alu-adjacent overmethylation in the body was not significantly different among H. pylori–positive controls, cancer patients, and dysplasia patients. Methylation of the two CpG-island gene types was further analyzed based on the level of gastric mucosal atrophy (Fig. 1B). Overmethylated Alu-adjacent genes in the body of cancer and dysplasia patients tended to be increased along the progression of gastric mucosal atrophy, involving one or more Alu-adjacent genes. LTR-adjacent genes were significantly overmethylated in the antrum and body of cancer patients with moderate or severe atrophy (p < 0.05), showing two or more overmethylated LTR-adjacent genes. They were also significantly overmethylated in the body of dysplasia patients with severe atrophy (p=0.020), but not with moderate atrophy.
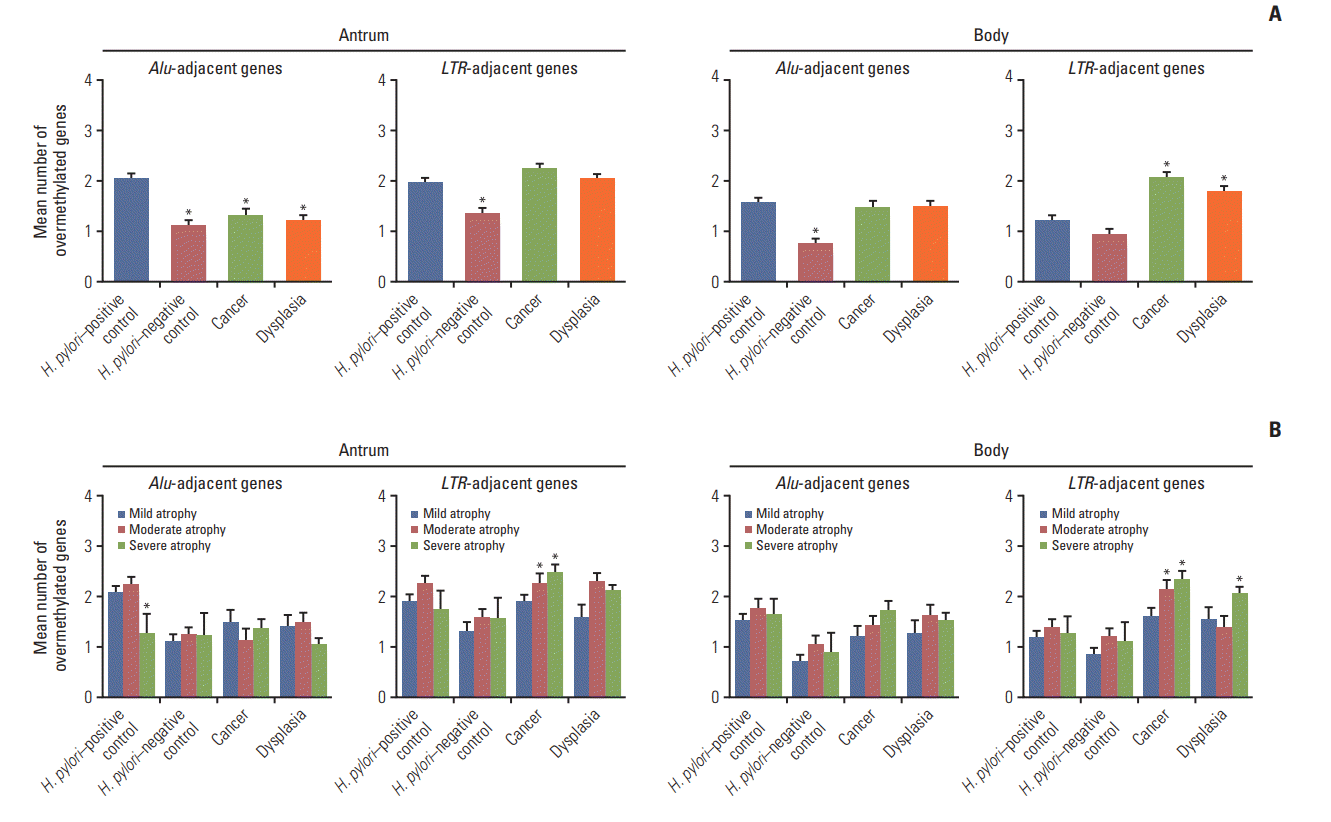
Fig. 1.
Comparison of numbers of overmethylated CpG-islandgenes among four subject groups: Helicobacter pylori–positive controls, H. pylori–negative controls, patients with gastric cancer, and patients with dysplasia. Numbers of overmethylated genes were separately calculated for CpG-island housekeeping genes adjacent to Alu retroelements (CDH1, ARRDC4, PPARG, and TRAPPC2L) or LTR retroelements (MMP2, CDKN2A, RUNX2, and RUNX3). (A) Numbers of overmethylated genes are represented as mean±standard error of mean (SEM) (*p< 0.05 compared to H. pylori–positive controls based on one-way ANOVA with Tukey’s post-hoc tests). (B) Numbers of overmethylated genes were further analyzed according to the level of gastric mucosal atrophy. They are presented as mean±SEM (*p< 0.05 compared to mild atrophic cases based on one-way ANOVA with Tukey’s post-hoc tests).
![]()
3. Age-related methylation of stomach-specific genes lacking CpG islands
TFF2, TFF1, PGC, ATP4B, and PGA are expressed in both the gastric antrum and body, whereas TFF3 in the antrum and GHRL in the body [14]. APC is inactive in both the antrum and body. Antrum-specific TFF3 and body-site specific GHRL were more methylated in the body and antrum respectively regardless of H. pylori infection (S6 Table). TFF2 methylation fluctuated with age showing three ambiguous peaks at ages 46, 55, and 60 years in the antrum of H. pyloripositive controls (Fig. 2A). TFF3 methylation showed two early peaks at ages 46 and 49 and a late peak at age 59. APC methylation tended to be decreased three times at ages 48, 54, and 59.
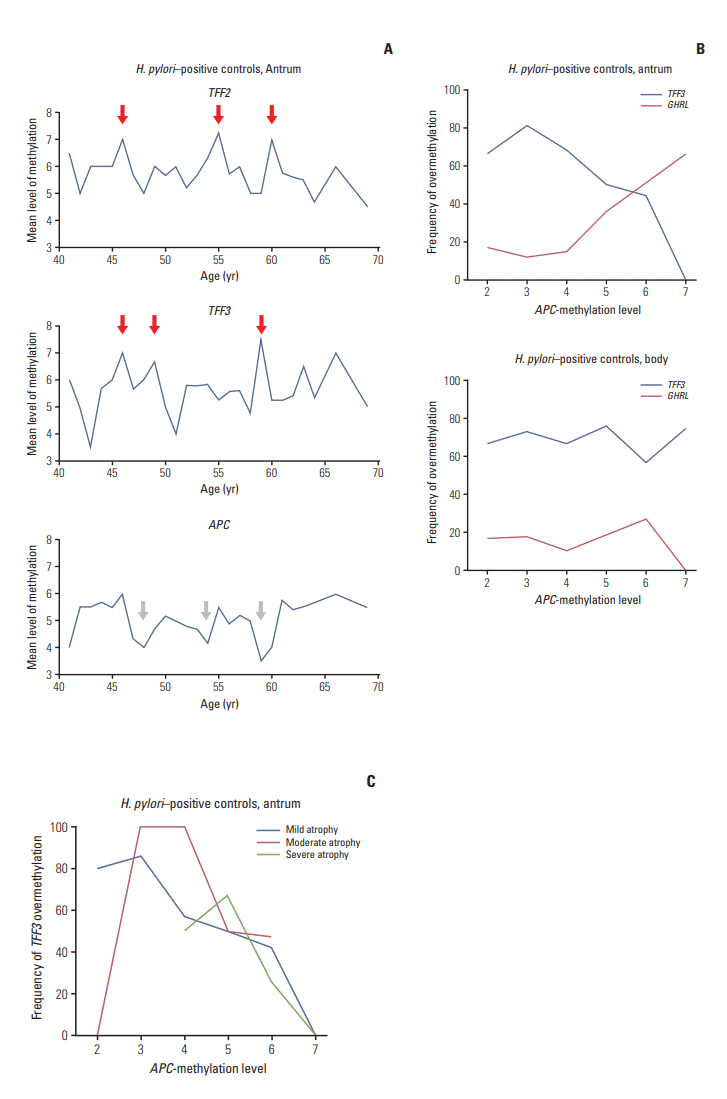
Fig. 2.
Analysis of age-related methylation in Helicobacter pylori–positive controls. (A) Age-related methylation changes of TFF2, TFF3, and APC were evaluated in the antrum by mean level of methylation estimated by 10-level classification. Open and closed vertical arrows indicate low trough and high peak respectively. (B) Age-related methylation of antrum-specific TFF3 and body-specific GHRL was evaluated in the antrum and body by the frequency of overmethylation. Age-related methylation curves were plotted as a function of age-dependent APC-methylation level. (C) Age-related methylation of TFF3 in the antrum was analyzed according to three levels of gastric mucosal atrophy.
![]()
Antrum-specific TFF3 was significantly overmethylated in the antrum of H. pylori–positive controls (p=0.003) (S5 Fig.). Inactive APC is known to be methylated in an age-dependent manner [14,15]. Age-related pattern of TFF3 and GHRL methylation was evaluated using APC methylation as a standard methylation age (Fig. 2B, S7 Fig.). In the antrum, TFF3 methylation peaked at APC methylation level 3 and GHRL methylation increased with increasing APC methylation levels. TFF3 methylation was further evaluated depending on the level of gastric mucosal atrophy in the antrum of H. pylori–positive controls (Fig. 2C). The frequency curve of TFF3 overmethylation reached an early peak at APC methylation level 3 in mild atrophic cases. The peak of TFF3 overmethylation frequency was shifted to APC methylation levels 3 and 4 in the antrum with moderate atrophy and to APC methylation 5 in severe atrophic cases. Consequently, TFF3 methylation peaked sequentially along the three levels of gastric mucosal atrophy.
The number of CpG-island overmethylation was slowly increased to a plateau over APC methylation levels 3 to 6 in both the antrum and body of H. pylori–positive controls (S8 Fig). The curve of Alu-adjacent overmethylation reached a higher plateau than that of LTR-adjacent overmethylation. The peak of Alu-adjacent methylation was ambiguous at the three levels of gastric mucosal atrophy in the body.
4. Relationship between level of gastric mucosal atrophy and age of overmethylation
H. pylori–positive controls examined in this study took an average of 14 years to develop from mild (mean age, 51±9 years) to severe (65±10 years) levels of gastric mucosal atrophy (Fig. 3A). H. pylori–positive controls with moderate atrophy (54±9 years) were three years older than those with mild atrophy (51±9 years). Similarly, Alu-adjacent overmethylation at moderate atrophic level (54±9 years) appeared two years later than that at mild atrophic level (52±9 years). The age difference of TFF3 overmethylation between mild (50±9 years) and moderate (57±9 years) atrophic cases was bigger than that of Alu-adjacent overmethylation.
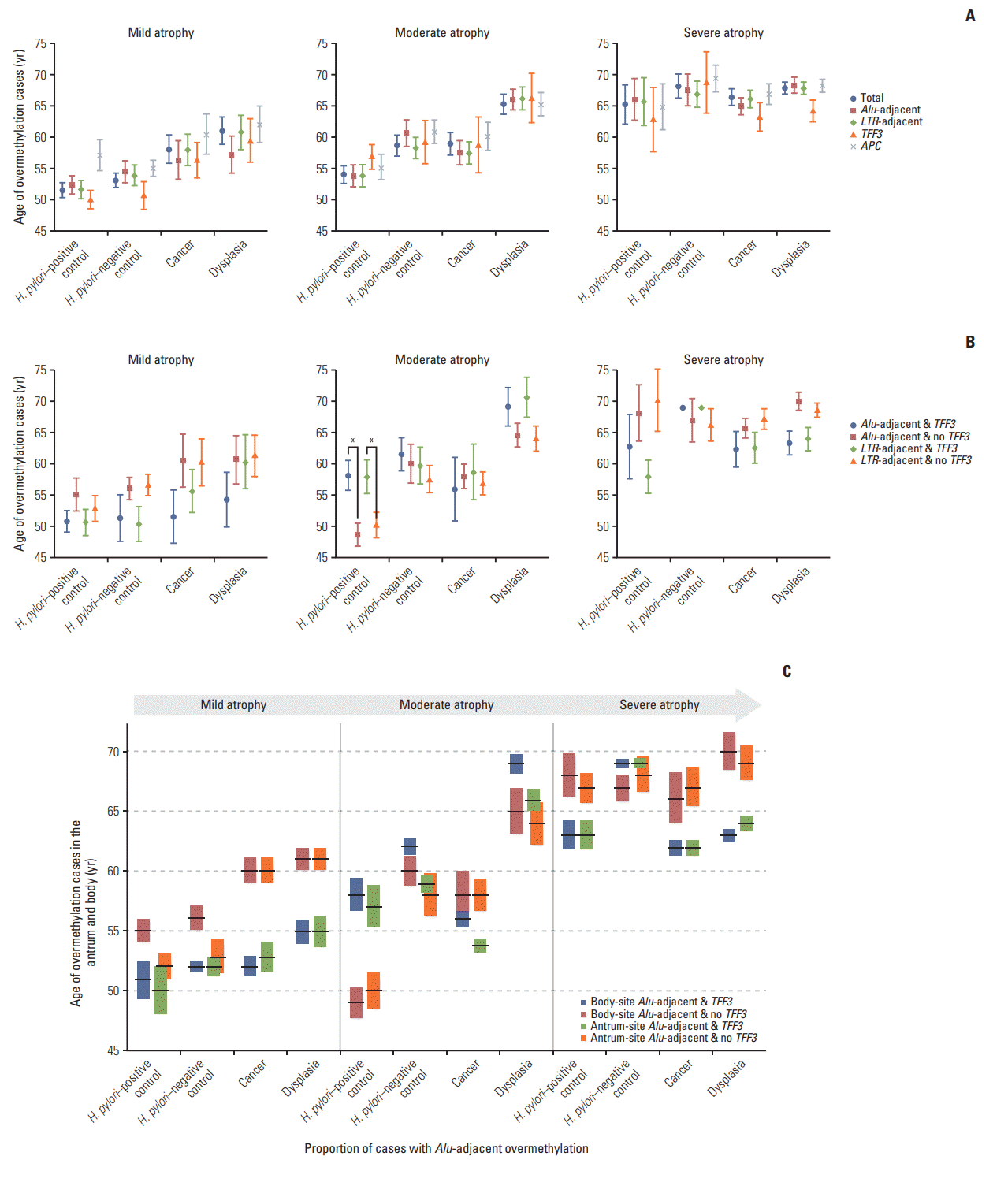
Fig. 3.
Overmethylation age of Alu-adjacent genes, LTR-adjacent genes, atrophy-sensitive TFF3, and age-dependent APC in four subject groups. Each of the four subject groups was subgrouped according to three atrophic levels. (A, B) Mean age of subjects showing overmethylated Alu- and LTR-adjacent genes in the gastricbody and overmethylated TFF3 and APC genes in the gastric antrum were analyzed. (A) Mean ages were compared between cases with Alu- or LTR-adjacent overmethylation and with TFF3 overmethylation. (B) Mean ages were compared between cases with Alu- or LTR-adjacent overmethylation with and without TFF3 overmethylation. Error bars indicate standard error of mean. *p< 0.05 by Student’s t test. (C) Changes in proportions of Alu-adjacent overmethylation during the progression of gastric atrophy. Cases of Alu-adjacent overmethylation with and without antrum-site TFF3 overmethylation were evaluated for both gastric antrum and body sites. The vertical length of bars represents the proportion (%) of overmethylation cases according to three atrophic levels in each subject group. A horizontal line across the proportion bar indicates the mean age of each methylation group.
![]()
Body-site CpG-island overmethylation and accompanying antrum-site TFF3 overmethylation were analyzed to determine the age of overmethylation that extended to the gastric body (Fig. 3B). In H. pylori–positive controls, TFF3+Alu overmethylation was found at similar intervals among mild (51±9 years), moderate (58±9 years), and severe levels (63±10 years) of mucosal atrophy. Especially, TFF3+Alu overmethylation in moderate atrophic cases was significantly old compared to Alu-alone overmethylation (58±9 vs. 49±7 years, p=0.005). There was also a significant difference in age between TFF3+LTR overmethylation and LTR-alone overmethylation (58±10 vs. 50±8 years, p=0.029). In gastric cancer patients with moderate atrophy, ages of TFF3+Alu overmethylation (56±12 years) and Alu-alone overmethylation (58±8 years) were similarly old. TFF3+Alu overmethylation was similarly old for severe mucosal atrophic cases including H. pylori–positive controls (63±10 years), cancer patients (62±9 years), and dysplasia patients (63±6 years).
The proportion and mean age of Alu-adjacent overmethylation and accompanying TFF3 overmethylation were depicted for both the antrum and the body (Fig. 3C). In H. pylori–positive controls, the proportion of TFF3+Alu overmethylation tended to be consistently high in the antrum and body of H. pylori–positive controls during the progression from mild to severe atrophy. The proportion of Alu-alone overmethylation in the body was increased from mild to severe levels of gastric mucosal atrophy in cancer patients (mild atrophy, 30%; moderate atrophy, 52%; and severe atrophy, 61%). Dysplasia patients with severe (45%) atrophy showed a higher proportion of Alu-adjacent overmethylation than those with mild atrophy (26%). Alu-alone overmethylation in moderate atrophic cases (antrum, 50±7 years; body, 49±7 years) was found as early as TFF3+Alu overmethylation in mild atrophic cases (antrum, 50±9 years; body, 51±9 years) in both the antrum and the body. The age of TFF3+Alu overmethylation in the antrum (57±9 years) and body (58±9 years) of moderate atrophic cases was similarly old.
5. Analysis of cancer-related methylation
The number of Alu-adjacent overmethylation was similar among H. pylori–positive controls, cancer patients, and dysplasia patients (Fig. 1A). Whereas, the number of LTR-adjacent overmethylation was higher in the body of cancer patients with moderate atrophy (Fig. 1B). The cancer patients demonstrated transitional-CpG overmethylation more frequently in the LTR-adjacent gene group than in the Aluadjacent gene group. LTR-dominant methylation type that was related with gastric cancer was defined as when the number of overmethylation involving at least one Alu-adjacent gene was higher in the LTR-adjacent gene group than that in the Alu-adjacent gene group. ORs and 95% CIs for the association between LTR-dominant methylation type and risk of gastric cancer or dysplasia were calculated (Table 2). LTR-dominant methylation type was more strongly associated with gastric cancer patients (OR, 5.2; 95% CI, 2.8 to 9.8) than with dysplasia patients (OR, 2.5; 95% CI, 1.3 to 4.8). According to atrophic level, the LTR-dominant methylation type was associated with cancer risk (OR, 5.3; 95% CI, 2.0 to 14.1), but not with dysplasia risk (OR, 2.4; 95% CI, 0.8 to 7.4) in moderate atrophic cases. It was frequently detected in both cancer patients (OR, 14.5; 95% CI, 4.3 to 49.1) and dysplasia patients (OR, 11.3; 95% CI, 3.3 to 38.1) with severe atrophy.
Table 2.
Adjusted odds ratios for the associations of LTR-dominant methylation type with gastric dysplasia or cancer
|
No. (%) |
OR (95% CI)a) | Cancer, n (%) | OR (95% CI)a) | ||
|---|---|---|---|---|---|
| Control | Dysplasia | ||||
| LTR-dominant methylation type | |||||
| Absence | 182 (89) | 82 (69) | 1.0 | 54 (55) | 1.0 |
| Presence | 23 (11) | 36 (31) | 2.5 (1.3-4.8) | 45 (45) | 5.2 (2.8-9.8) |
| LTR-dominant methylation type stratified by level of gastric mucosal atrophy | |||||
| Absence, mild | 88 (43) | 20 (17) | 1.0 | 21 (21) | 1.0 |
| Absence, moderate | 62 (30) | 22 (18) | 1.2 (0.6-2.6) | 16 (16) | 1.0 (0.5-2.1) |
| Absence, severe | 16 (8) | 40 (34) | 4.8 (2.1-10.9)b) | 17 (17) | 2.8 (1.1-6.8)b) |
| Presence, mild | 9 (4) | 3 (3) | 1.3 (0.3-5.9) | 6 (6) | 2.9 (0.9-9.6) |
| Presence, moderate | 10 (5) | 7 (6) | 2.4 (0.8-7.4) | 15 (15) | 5.3 (2.0-14.1)b) |
| Presence, severe | 4 (2) | 26 (22) | 11.3 (3.3-38.1)b) | 24 (25) | 14.5 (4.3-49.1)b) |
![]()
Discussion
H. pylori infection induces a various number of methylation-inducible mucosal cells from individual to individual. Despite the cyclic replacement of glandular stem cells [7], few studies have demonstrated cyclic methylation peaks. TFF2 is strongly expressed in both the antrum and body, playing a master role in induction of stem cell trans-differentiation [14,17]. Strong expression of this master gene made no increase in H. pylori–associated methylation. Antrum-specific TFF3 and body-specific GHRL were undermethylated in the antrum and body, respectively [14]. This is in accordance with an inverse relationship between transitional-CpG methylation and gene expression [23]. Strong expression of master genes is organized by priority to reestablish new stem cell phenotypes and to lead stem cell repopulation [17]. The tissue-specific methylation can periodically fluctuate under the influence of gene expression after the establishment of new stem cell phenotypes [24]. The results of stomach-specific gene methylation might represent the cyclic repopulation of stem cells under the influence of gene expression rather than by accidental events.
Housekeeping genes containing CpG islands made slow methylation curves. This suggests that CpG-island genes keep their overmethylation for a long time in order to stabilize newly established cell phenotypes. H. pylori–associated TFF3 methylation was found to mark three overmethylation peaks at three levels of gastric mucosal atrophy (Fig. 2C). Rapid TFF3 overmethylation was found earlier than Alualone overmethylation in H. pylori–positive controls with mild atrophy, indicating an early stage of overmethylation. The age of TFF3+Alu overmethylation appeared at similar intervals among mild (51 years), moderate (58 years), and severe (63 years) atrophic cases. Whereas, the age of moderate atrophic cases (54 years) was close to that of mild atrophic cases (52 years). The age of stomach-specific gene methylation is thought to represent cyclic methylation changes at certain times.
H. pylori–positive controls with moderate mucosal atrophy demonstrated TFF3 overmethylation existing in a wide range of APC methylation (Fig. 2C). This implies that the moderate atrophic cases involve several methylation cycles. There was a significant difference in the mean age between early Alualone overmethylation and late TFF3+Alu overmethylation in the moderate mucosal atrophic cases (Fig. 3B). H. pylori–positive controls with moderate mucosal atrophy appeared to include two distinct cycles of transitional-CpG methylation, each of which resulted from early-onset and late-onset mucosal atrophy. Early-onset moderate mucosal atrophy is likely to impede TFF3 overmethylation at primary methylation cycle. Meanwhile, late TFF3+Alu overmethylation at the mean age of 58 years presents early overmethylation in cases of late-onset mucosal atrophy. The age of early Alu-alone overmethylation (49 years) in moderate atrophic cases was comparable with that of TFF3+Alu overmethylation in mild atrophic cases (51 years). This suggests that the cyclic repopulation of stem cells is initiated at a certain period regardless of the level of gastric mucosal atrophy.
LTR-adjacent overmethylation not Alu-adjacent overmethylation was significantly frequent in cancer patients (Fig. 1). In cancer patients, early-onset atrophic changes, inducing a high number of methylation-inducible cells, might fail to increase Alu-adjacent methylation. Indeed, previous studies on Alu retroelements have demonstrated hypomethylation changes in H. pylori-infected subject with severe atrophy and patients with gastric cancer [25]. Alu-adjacent overmethylation was found to be later in gastric cancer patients with moderate atrophy (58 years) than in H. pylori–positive controls with mild (51 years) or moderate atrophy (49 years) (Fig. 3B). The Alu-adjacent CpG-island genes can produce Alu RNAs to quickly stabilize cell phenotypes prior to transitional-CpG methylation [11,17]. There appears to be a window of Alu RNA-dependent stabilization during the expansion of new stem cells. Cancer-risk moderate atrophy may prolong the Alu RNA-dependent step of phenotypic stabilization for a cycle of glandular cell repopulation. The risk of gastric cancer seems to be initiated with impeding phenotype-stabilizing methylation of new stem cells. LTR-adjacent overmethylation was frequent in both cancer and dysplasia patients at a severe level of gastric mucosal atrophy (Fig. 1B). Methylation-inducible cells are likely to transform into cancerous cells and subsequently into dysplastic cells.
The proportion of TFF3+Alu overmethylation was low in gastric cancer (20%) and dysplasia (17%) patients with severe atrophic cases (Fig. 3C). However, the proportion of Aluadjacent overmethylation tended to be increased in both cancer and dysplasia patients (Fig. 3C). Such increased methylation of CpG-island genes can change the epigenetic status of new stem cells from unstable to stable during subsequent cell repopulation. Gastric mucosa appears to be at a relatively low risk for cancer in severe atrophic cases, even though neoplastic transformation into both gastric cancer and dysplasia occur frequently in such cases.
CpG-island methylation was not increased in dysplasia patients with moderate atrophy (66 years) who were older than gastric cancer patients (58 years) (Figs. 1B and 3A). This suggests that weak H. pylori infection introduces late-onset slow-progressing gastric mucosal atrophy, staying at a moderate level, and leads to a low number of methylationinducible cells.
In previous prospective studies, intense early-onset H. pylori infection has been found to be able to elevate the risk of gastric cancer, which will induce severe mucosal atrophy eventually [1,3]. Both cancer (66 years) and dysplasia (68 years) patients with severe atrophy showed frequent LTRadjacent overmethylation at similar ages (Figs. 1B and 3A). It is likely that early-onset moderate atrophy progresses rapidly toward severe atrophy. The window period for developing gastric cancer is predictable based on the following sequence of H. pylori-associated events: intense H. pylori infectionearly-onset rapid-progressing moderate atrophy impeded Alu-adjacent methylation and increased LTRadjacent methylationsevere atrophy.
Cancer risk of gastric mucosal atrophy has been assessed previously by endoscopic recognition of the atrophic border and by the Sydney system including histologic examination of biopsies [21,26]. There are good agreements between the two atrophy-staging systems. A prospective study on gastric cancer occurrence, involving 5,373 subjects (mean age, 50 years) over 10 years has reported that the relative risk of gastric cancer is the greatest among subjects with moderate atrophy [27]. The peak value of relative risk is reached after 4-6 years of follow-up, followed by attenuation with age and severe atrophic changes. These findings are in accordance with our results showing that rapid-progressing atrophy impeding methylation (mean age, 58 years) is followed by severe atrophy with a low risk of gastric cancer. However, atrophy-staging systems provide little practical information at the time of endoscopic examination because neither endoscopic findings nor histological parameters are available for the identification of rapid-progressing moderate atrophy. On the other hand, cancer-related methylation type associated with an increased risk of gastric cancer can be useful for highrisk individual detection at a moderate level of gastric mucosal atrophy (Table 2).
Previous studies on H. pylori-associated methylation have mainly concentrated on weakly methylated CpG-island centers without considering transitional-CpG sites [18]. There are considerable inter-observer differences on the association of hypermethylated CpG-island centers with inactivation of tumor suppressor genes (S9 Table) [28]. When the type of retroelements adjacent to genes examined in previous studies was analyzed, most CpG islands were classified as Alu retroelements. Only a small number of CpG islands were classified as LTR retroelements or no retroelement. Among six studies on the CpG-island centers of LTR-adjacent CDKN2A, two displayed a higher level of methylation in cancer patients than that in H. pylori-positive controls. However, this was not observed in the other four studies. These inconsistent results might be due to weak methylation of the CpGisland centers that show a lack of reproducibility with different detection methods and various arbitrary cutoff points. The transitional-CpG sites displayed a wide range of methylation variations compared with the CpG-island centers in cell-differentiation and human gastric cancer studies [19,20]. In addition, site-specific methylation changes were clearly observed in the transitional-CpG sites, but not in the CpGisland centers [15,18]. Therefore, the transitional-CpG sites are profitable for clarifying H. pylori-associated methylation (Fig. 1A).
A phenotypic shift from undifferentiated to differentiated types is clearly observed in both intestinal metaplasia and gastric cancer [12,13]. These findings imply that new gastric stem cells at an early adaptation stage are prone to restore properties of mesenchymal cells. So the possibility of metaplastic or neoplastic differentiation reaches a peak at an unstable step of phenotypic stabilization, suggesting that both intestinal metaplasia and gastric cancer appear as a paracancerous phenomenon, not a precancerous cascade [29]. Gastric dysplasia may also result from a paracancerous phenomenon. Gastric cancer is closely associated with highgrade dysplasia compared with low-grade dysplasia [30]. Morphological similarities of gastric dysplasia and cancer implicate a cause-and-effect relationship between gradual phenotypic stabilization and aberrant differentiation rather than a dysplasia-cancer sequence. Accordingly, gastric dysplasia might occur earlier or later than gastric cancer for the period of impeded CpG-island methylation.
In conclusion, moderate atrophy of the gastric mucosa impedes phenotype-stabilizing Alu-adjacent methylation for nine years (a cycle of glandular stem cell repopulation) in gastric cancer patients, but not in dysplasia patients. The risk of gastric dysplasia could arise later than that of gastric cancer during phenotypic stabilization which grows with transitional-CpG methylation. The window period for developing gastric cancer may be predicted by analyzing the age of transitional-CpG methylation and the level of gastric mucosal atrophy.
 XML Download
XML Download