

 PDF
PDF Citation
Citation Print
Print


Introduction
The incidence rate of colorectal cancer (CRC) is high in males compared with females, regardless of age, ethnicity, and geographic regions [1]. Epidemiologic studies have shown an obvious decrement of CRC incidence with oral contraceptive use [2], and lowered risk of CRC with estradiol plus progestin therapy [3]. Some preclinical research has yielded conflicting results on the influence of female sex hormones on CRC. That is, in ovariectomized female C57BL/6J mice with a germ-line APC gene mutation, intestinal adenomas were reportedly increased by 77 % (p < 0.05) compared to non-ovariectomized females, while supplementation of 17β-estradiol to ovariectomized female C57BL/6J mice reduced the number of adenomas to the same level as non-ovariectomized mice [4]. However, in an APCPirc/+ rat model, ovariectomized rats did not develop a higher prevalence of adenomas, while orchidectomy protected against colonic tumorigenesis [5]. In our previous study, we suggested the protective roles of estradiol in colorectal tumorigenesis by showing more tumor multiplicities in azoxymethane and dextran sulphate sodium (AOM/DSS)-treated male mice compared to AOM/DSS-treated female mice [6].
Estradiol increased nuclear factor erythroid 2-related factor 2 (Nrf2) activity in breast cancer cell line [7]. Yet in CRC, there is no comprehensive knowledge about estradiol as an upstream regulator of Nrf2. Protein kinase Cδ (PKCδ), an important mediator in the Gα13 signaling pathway, promotes Nrf2 activity [8]. In addition, PKC has been closely related to the protective effect of estradiol on vascular reactivity after shock in female rats [9]. Furthermore, estradiol-induced protein synthesis in mouse uterine epithelial cells was also mediated through the PKC signaling pathway [10]. Estradiol increased the mRNA level of PKCδ in the colonic epithelium of rats [11]. Thus, the Gα13-PKCδ signaling pathway could be an upstream regulator of Nrf2 in CRC, and estradiol might play a role in this cascade.
The enhancement of colitis-associated CRC development in Nrf2-deficient mice treated with AOM/DSS [12] supports the protective influence of Nrf2 against colonic inflammation. There are several suggested mechanisms of the Nrf2-mediated prevention of inflammation and tumorigenesis. First, the activation of Nrf2 and cross-talk between Nrf2 and nuclear factor-κB (NF-κB) downregulate pro-inflammatory signaling by suppressing NF-κB directly [13]. Second, Nrf2 is one of the most essential transcription factors that regulate the expression of anti-oxidant enzymes [14]. Lastly, the close relationship of Nrf2 with the activating mechanism of the Nod-like receptor protein 3 (NLRP3) inflammasome was recently reported [15]. Caspase-1 activated by NLRP3 inflammasome triggers pyroptosis [16], and pyroptosis might elicit an anti-cancer immune reaction [17].
Inflammation is an important factor in the pathophysiology of colitis-associated and sporadic CRC. For example, Saleiro et al. [18] demonstrated the higher levels of inflammatory cytokines and polyp development at weeks 9 and 16 in AOM/DSS-treated estrogen receptor β (ERβ) knockout mice, compared to wild-type mice. However, almost no studies have thoroughly evaluated the early inflammation stage of tumorigenesis, since in most of the studies, the animals were sacrificed after adenoma formation.
From this background, we hypothesized that the observed sex difference in CRC incidence may be due to estradiol-mediated down-regulation of inflammation, which might somehow affect the CRC cascade. To explore this hypothesis, we assessed the temporal role of Nrf2 in modulating inflammation and carcinogenesis through the regulation of the NF-κB–mediated pro-inflammatory pathway, anti-oxidant enzymes, and the NLRP3 inflammasome.
Materials and Methods
1. Animals
Four-week-old male and female ICR mice (Orient Co., Ltd., Seoul, Korea) were housed in cages, and maintained at 23°C with a 12/12-hour light/dark cycle under specific pathogen-free conditions.
2. Experimental design
Fig. 1A shows the experimental design. After 1 week of acclimatization, male and female mice were randomized into five groups (n=20-36/group). Group 1 male control (M-con) mice were sacrificed at week 2 (n=4), and weeks 10 and 16 (n=6 each). Group 2 comprised male mice treated with AOM/DSS (M-AOM/DSS). The mice were sacrificed at week 2 (n=6), and at weeks 10 and 16 (n=12 each). Group 3 comprised AOM/DSS-treated male mice administered estradiol (M-AOM/DSS+estr). The mice were sacrificed at week 2 (n=6), and weeks 10 and 16 (n=12 each). Group 4 comprised female control mice (F-con). They were sacrificed at week 2 (n=4), and weeks 10 and 16 (n=6 each). Group 5 comprised AOM/DSS-treated female mice (F-AOM/DSS). They were sacrificed at week 2 (n=6), and weeks 10 and 16 (n=12 each). AOM/DSS-treated male and female mice were intraperitoneally injected with AOM (10 mg/kg; Sigma-Aldrich, St. Louis, MO) on day 0 in the experimental schedule. For induction of colitis, 2.5 % (w/v) DSS (MP Biomedicals, Aurora, OH) was supplied in drinking water for 7 days, 1 week following the injection of AOM [6]. The quantity of DSS consumed in the drinking water was checked on days 7, 9, and 11. M-AOM/DSS+estr mice were intraperitoneally injected each day for 7 days with 17β-estradiol (10 mg/kg; Sigma-Aldrich) dissolved in olive oil. The injections were done during the same period of DSS consumption. Animals were euthanized by CO2 asphyxiation at 2, 10, and 16 weeks after AOM injection (Fig. 1A).
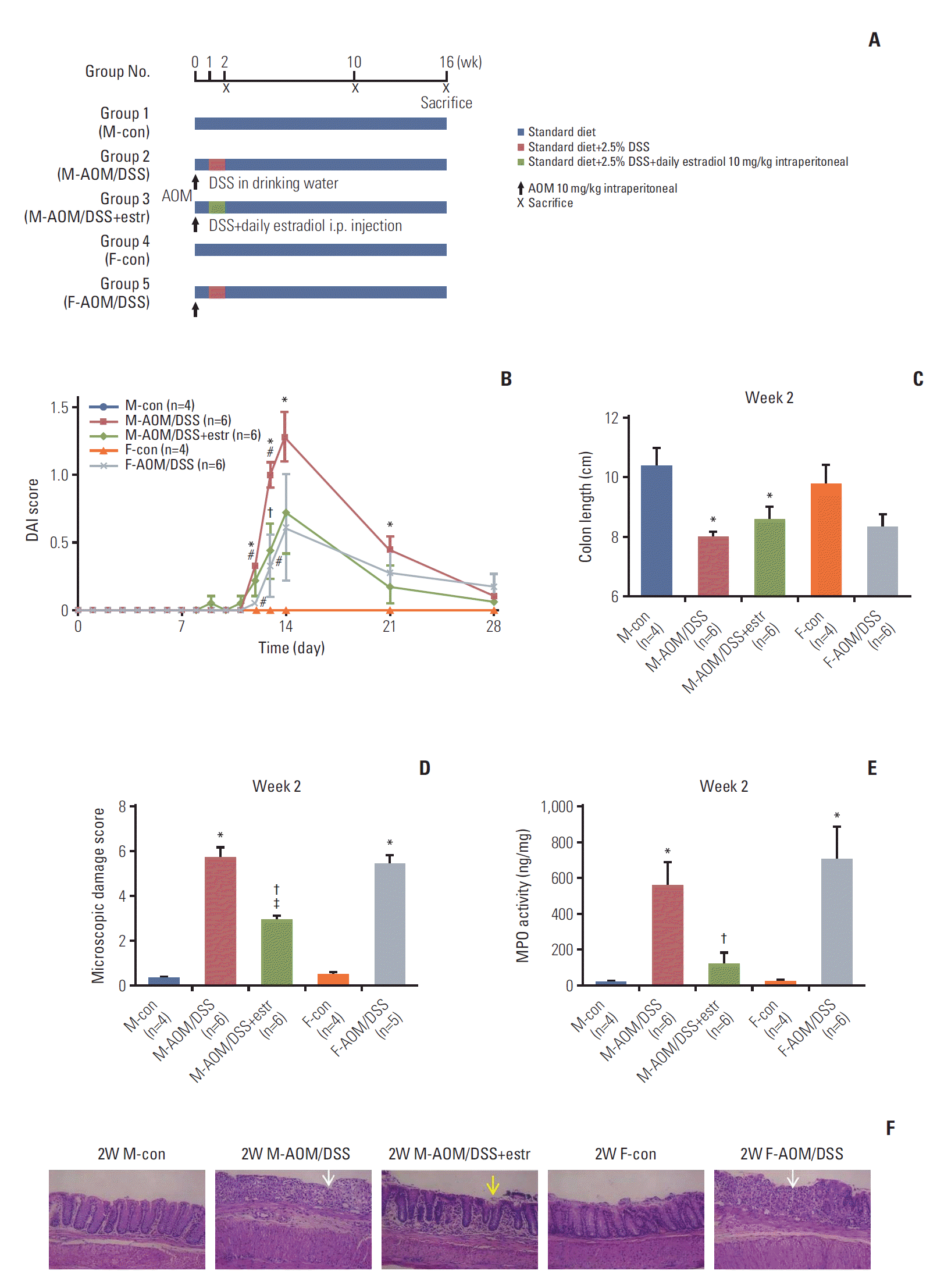
Fig. 1.
Estradiol prevents wasting disease progression in azoxymethane/dextran sulfate sodium (AOM/DSS)–induced colitis. (A) Scheme for the experimental course of AOM/DSS promoted colitis-associated tumorigenesis. The mice were injected AOM on day 0. DSS in drinking water (2.5%) and estradiol supply was provided from day 7 to 13. Mice were sacrificed at week 2, 10, and 16. (B) Disease Activity Index (DAI) was decreased by estradiol. (C) Colon length at week 2. (D) Macroscopic damage score at week 2. (E) Myeloperoxidase (MPO) activity in colonic tissues at week 2. (F) Histopathologic findings of the colonic mucosa (H&E staining, ×200) at week 2. In control mice, the mucosa is normal in males and females. However, near-total crypt loss and infiltration of severe inflammatory cell of colonic mucosa (white arrow) are seen in both males and females. Estradiol treatment significantly decreased histologic damage, with only mild erosion (yellow arrow). *p < 0.05 compared to control, †p < 0.05 compared to AOM/DSS group, ǂp < 0.05 between estradiol-treated group and female AOM/DSS group, #p < 0.05 between the male AOM/DSS group and the female AOM/DSS group. M, male; F, female; estr, estradiol.
![]()
3. Evaluation of clinical symptoms
Clinical symptoms were evaluated using the Disease Activity Index (DAI), which includes body weight loss, stool characterization, and hematochezia [6]. DAI was scored by two technicians (L.H.N. and D.C.) in a blinded manner.
4. Lesion enumeration
Colons extracted from cecum to the rectum were opened longitudinally, and stool was washed out with phosphate-buffered saline. Colon length was measured from cecum to rectum using a ruler. Polypoid lesions with a diameter < 2 mm or > 2 mm were independently counted by three gastroenterologists in a blinded manner. Tumor multiplicity was defined as the number of gross polyps approved by the three gastroenterologists.
5. Tissue processing, histopathology, and immunohistochemical analysis
After extraction from the peritoneum, the colon was divided into proximal and distal portions. The proximal colon was half of the colon to 1.5 cm distal from the ileocecal valve. The distal colon was the other half up to the rectum 1.5 cm from the anal verge. One or two representative polyps of each sample were prepared for histological analysis. These samples were fixed with phosphate-buffered formalin, and stained with hematoxylin and eosin. Other portions were frozen in lipid nitrogen, and kept at –70°C, until use in the biochemical assays. The tumor incidence (%) was determined as the percentage of rats having more than one tumor. The classification of adenoma and adenocarcinoma was performed as previously described [6]. The depth of invasion by adenocarcinoma in the colonic tissues was specified as mucosa or submucosa [6,19]. Their incidence was also measured.
Immunohistochemical (IHC) analysis of Nrf2 was performed. Tissue sections were treated with 3% hydrogen peroxide, and nonspecific binding sites were blocked. The sections were incubated with anti-Nrf2 antibodies (ab31163, Abcam, Cambridge, MA). An automatic immunostainer (BenchMark XT, Ventana Medical Systems, Tucson, AZ) and UltraView Universial DAB detection kit (Ventana Medical Systems) were used for immunostaining. The proportion of the number of immune-stained in total cells of all crypts were calculated.
6. Scoring of microscopic damage
Histological severity was assessed using microscopic damage score reflecting colonic epithelial damage and depth of infiltration with inflammatory cells as previously described [6]. This was evaluated by a pathologist (E.S.) in a blinded manner.
7. Measurement of inflammatory cytokines
The levels of myeloperoxidase (MPO) in the colonic tissues were examined by ELISA (R&D Systems, Minneapolis, MN). Every assay was performed in triplicate.
8. Western blot analysis
Protein extracts were isolated using RIPA buffer (Cell Signaling Technology, Beverly, MA). Cytoplasmic and nuclear lysates were separated using a NE-PER Nuclear Cytoplasmic Extraction Reagent kit (Pierce, Rockford, IL), according to the manufacturer’s instructions. Protein concentration was determined using the BCA protein assay reagent (Pierce). Protein samples were separated by 8 to 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis. After blocking, membranes were incubated overnight at 4°C with specific antibodies. S1 Table of the Supporting Information (SI) lists the primary antibodies in detail. Horseradish peroxidase–conjugated anti-rabbit, anti-goat, or anti-mouse immunoglobulin (Santa Cruz Biotechnology, Dallas, TX) was used as secondary antibodies.
9. Quantitative real-time PCR analysis
RNA was isolated from colon tissue using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruments, and quantified using a NanoDrop ND-1000 device (Thermo Scientific, Wilmington, DE). cDNA was synthesized using the High Capacity cDNA reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed using Power SYBR Green PCR Master mix and a Viia7 instrument (Applied Biosystems). The transcript levels of glyceraldehydes-3-phosphate dehydrogenase were used for sample normalization. S2 Table of the SI lists the primer sequences.
10. Statistical analyses
Data are expressed as mean±standard error of mean. Statistical significance was examined by Mann-Whitney test or Fisher exact test. A p-value of < 0.05 was considered to indicate statistical significance. All statistical analyses were conducted using SPSS ver. 18.0 (SPSS Inc., Chicago, IL) and GraphPad Prism software (GraphPad, La Jolla, CA).
11. Ethical statement
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University Bundang Hospital (BA1310-139/091-01). The procedures were in accordance with the Animals in Research: Reporting In Vivo Experiments (ARRIVE) statement.
Results
1. Estradiol ameliorates histologic evidence of colonic inflammation by week 2
We first analyzed DAI score, colon shortening, and severity of colitis, to evaluate the early impacts of estradiol. The M-AOM/DSS and F-AOM/DSS groups displayed higher DAI scores compared to the control mice (M-con and F-con groups), suggesting the induction of severe colitis (p=0.007 at week 2 for M-AOM/DSS group vs. M-con group) (Fig. 1B). The M-AOM/DSS+estr group had lower DAI scores than the M-AOM/DSS group on day 13 (p=0.049), and at week 2 (Fig. 1B). Colon length of the M-AOM/DSS group was shortened by inflammation at week 2 (Fig. 1C). Representative histologic images (Fig. 1F) and microscopic damage score revealed significantly less infiltration of inflammatory cells and mild cryptic damage for the M-AOM/DSS+estr group compared to the M-AOM/DSS group at week 2 (p=0.004 for microscopic damage score) (Fig. 1D). The M-AOM/DSS+estr group displayed a lower level of MPO, a mediator associated with intestinal inflammation, compared to the M-AOM/DSS group (p=0.020) (Fig. 1E). The collective data indicate that estradiol reduced the severity of DSS-induced colitis.
2. Estradiol attenuates colitis-associated, histology-evident tumorigenesis at weeks 10 and 16
Prominent polyps developed at weeks 10 and 16, mostly in the distal part of the colon (Table 1, Fig. 2A and B), consistent with a previous report [20]. The development of polyps was obvious in the M-AOM/DSS group, while only a few polyps developed in the F-AOM/DSS group at week 10 (p=0.014 for tumor number) (Fig. 2A and C). The findings provided evidence of a significant sex difference in colitisassociated tumor development. An astonishing result was the absence of visible polyps in the M-AOM/DSS+estr group at week 10 (Fig. 2A). The sex difference was also present at week 16; the F-AOM/DSS group displayed fewer tumors and lower incidence of colonic neoplasms compared to the M-AOM/DSS group (p=0.001 for tumor incidence) (Table 1, Fig. 2B and D). In the F-AOM/DSS group, CRC had developed in four of 12 (25%) of the mice at week 16, which was significantly lower than the M-AOM/DSS group (Table 1). Polyps were observed in the M-AOM/DSS+estr group at week 16, but at markedly fewer numbers than the M-AOM/DSS group (p=0.020) (Fig. 2B). These data were evidence of the protection conferred by both endogenous and exogenous estradiol against colitis-associated tumorigenesis. Data provided in Table 1 summarizes the microscopic incidence of colonic neoplasms. Adenocarcinoma that developed in the M-AOM/DSS group tended to have invasive growth, while that in the M-AOM/DSS+estr group displayed an intact muscularis mucosa lining at week 10 (Fig. 2C). The M-AOM/DSS group presented the most severe histological invasiveness at week 16 (Fig. 2D).
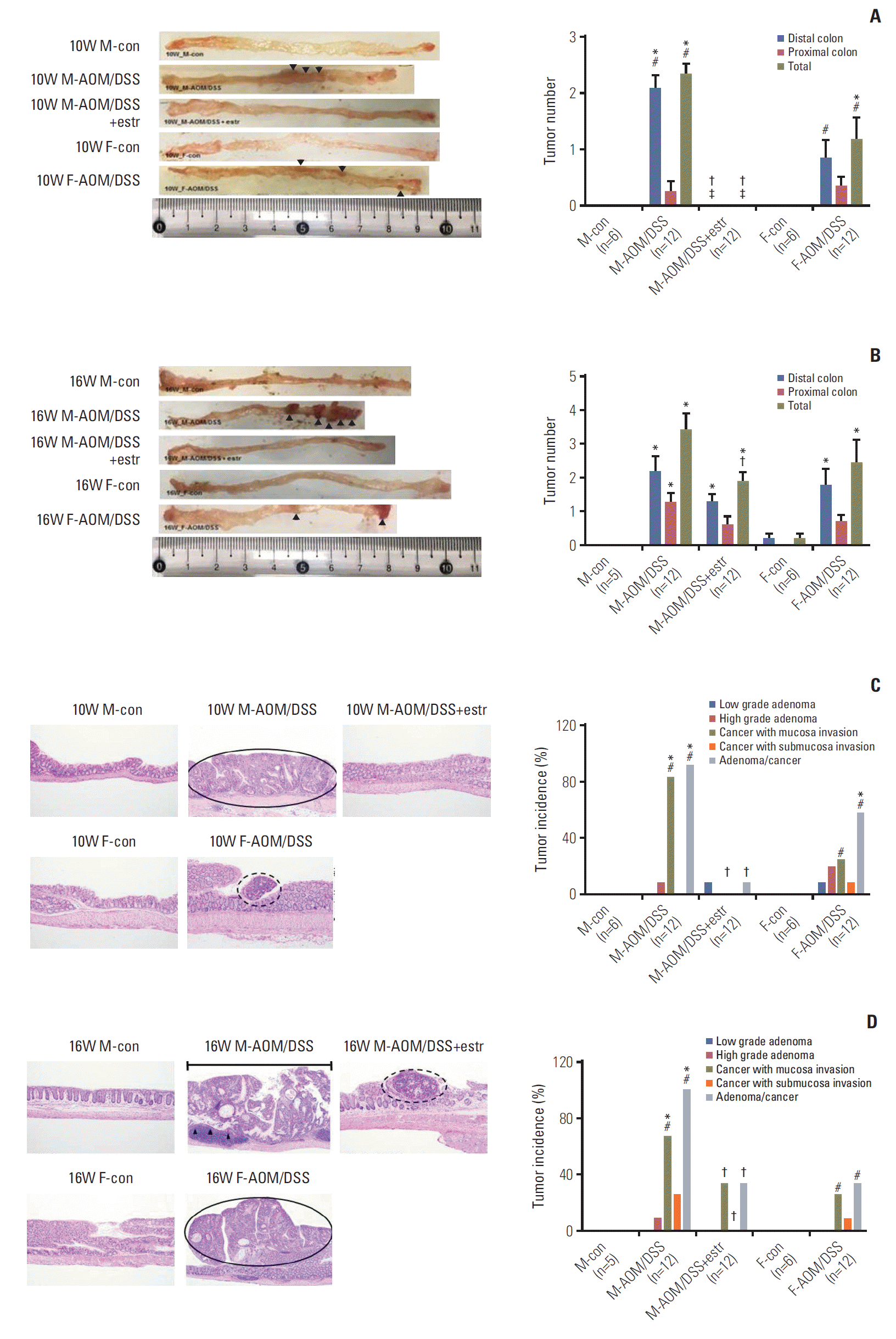
Fig. 2.
Effect of estradiol and sex-associated differences in the multiplicity of colorectal cancer at weeks 10 and 16. Macroscopic view (left panel) and multiplicity of the colons (right panel) in each group sacrificed at weeks 10 (A) and 16 (B). Arrowheads indicate the macroscopic polyps. Representative histological images at weeks 10 (C) and 16 (D). Adenoma is indicated with dashed line circle, adenocarcinoma with full line circle and a bar, and submucosal invasion with arrowheads. Quantification of invasion and incidence of cancer in each group at 10 and 16 weeks obtained by microscopic evaluation of the colonic tissues (H&E staining, ×100). *p < 0.05 compared to control, †p < 0.05 compared to the in azoxymethane/dextran sulfate sodium (AOM/DSS) group, ǂp < 0.05 between the estradiol-treated group and the female AOM/DSS group, #p < 0.05 between the male AOM/DSS group and the female AOM/DSS group. M, male; F, female; estr, estradiol.
![]()
Table 1.
Incidence and multiplicity of adenoma and cancer in colon
| Group | Low grade adenoma incidence | High grade adenoma incidence | Cancer with mucosa invasion | Cancer with submucosa invasion | Adenoma/cancer incidence | Adenoma/cancer multiplicity |
|---|---|---|---|---|---|---|
| 4-Week male | ||||||
| Control (n=4) | 0/4 (0) | 1/6 (16.7) | 0/4 (0) | 0/4 (0) | 0/4 (0) | 0.0 |
| AOM/DSS (n=6) | 1/6 (16.7) | 0/4 (0) | 0/6 (0) | 0/6 (0) | 1/6 (16.7) | 0.17±0.17 |
| AOM/DSS+E2 (n=6) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0.17±0.17 |
| p-valuea) | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 0.762 |
| p-valueb) | 1.000 | 1.000 | 1.000 | 1.000 | 0.455 | 1.000 |
| p-valuec) | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 0.523 |
| 4-Week female | ||||||
| Control (n=4) | 0/4 (0) | 0/4 (0) | 0/4 (0) | 0/4 (0) | 0/4 (0) | 0.0 |
| AOM/DSS (n=6) | 0/6 (0) | 1/6 (16.7) | 0/6 (0) | 0/6 (0) | 1/6 (16.7) | 0.33±0.21 |
| p-valuea) | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 0.221 |
| 10-Week male | ||||||
| Control (n=6) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0.0) | 0.0 |
| AOM/DSS (n=12) | 0/12 (0) | 1/12 (8.3) | 10/12 (83.3) | 0/12 (0) | 11/12 (91.6) | 2.33±0.19 |
| AOM/DSS+E2 (n=12) | 1/12 (8.3) | 0/12 (0) | 0/12 (0) | 0/12 (0) | 1/12 (8.3) | 0.0 |
| p-valuea) | 1.000 | 1.000 | 0.002* | 1.000 | < 0.001* | < 0.001* |
| p-valueb) | 1.000 | 1.000 | < 0.001* | 1.000 | < 0.001* | < 0.001* |
| p-valuec) | 1.000 | 1.000 | 0.012* | 1.000 | 0.155 | 0.014* |
| 10-Week female | ||||||
| Control (n=6) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0.0 |
| AOM/DSS (n=12) | 1/12 (8.3) | 2/12 (16.6) | 3/12 (25) | 1/12 (8.3) | 7/12 (58.3) | 1.17±0.39 |
| p-valuea) | 1.000 | 0.529 | 0.515 | 1.000 | 0.038* | 0.025* |
| 16-Week male | ||||||
| Control (n=6) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0.0 |
| AOM/DSS (n=12) | 0/12 (0) | 1/12 (8.3) | 8/12 (66.6) | 3/12 (25) | 12/12 (100) | 3.42±0.50 |
| AOM/DSS+E2 (n=12) | 0/12 (0) | 0/12 (0) | 4/12 (33.3) | 0/12 (0) | 4/12 (33.3) | 1.83±0.34 |
| p-valuea) | 1.000 | 1.000 | 0.013* | 0.515 | < 0.001* | 0.001* |
| p-valueb) | 1.000 | 1.000 | 0.220 | 0.217 | 0.001* | 0.020* |
| p-valuec) | 1.000 | 1.000 | 0.100 | 0.590 | 0.001* | 0.243 |
| 16-Week female | ||||||
| Control (n=6) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0/6 (0) | 0.0 |
| AOM/DSS (n=12) | 0/12 (0) | 0/12 (0) | 3/12 (25.0) | 1/12 (8.3) | 4/12 (33.3) | 2.42±0.71 |
| p-valuea) | 1.000 | 1.000 | 0.526 | 1.000 | 0.245 | 0.035* |
![]()
3. Effect of estradiol during colitis and cancer progression in terms of NF-κB
To further evaluate the estradiol effects on inflammatory factors at the molecular level, we measured NF-κB and its related pro-inflammatory enzymes, cytokines, and genes. First of all, we determined the expression levels of NF-κB by Western blot analysis at week 2. The M-AOM/DSS group had higher levels of NF-κB, compared to both the F-AOM/DSS and M-AOM/DSS+estr groups (Fig. 3A). Consistent with the NF-κB results, the levels of two major pro-inflammatory enzymes (cyclooxygenase-2 [COX-2] and inducible nitric oxide synthase [iNOS]), which are mainly regulated by NF-κB, were higher in the M-AOM/DSS group, compared to the F-AOM/DSS group (p < 0.05 for iNOS) (Fig. 3A). The protein and mRNA levels of NF-κB–related pro-inflammatory enzymes were decreased in the M-AOM/DSS+estr group compared to the M-AOM/DSS group at week 2, to similar levels of control mice (p < 0.05 for COX-2) (Fig. 3A and B). Next, we measured NF-κB–related pro-inflammatory cytokines (i.e. interleukin 6 [IL-6] and tumor necrosis factor α [TNF-α]) in the colonic mucosa at week 2 by real-time quantitative reverse transcription PCR (qRT-PCR). The F-AOM/DSS group displayed reduced levels of the pro-inflammatory cytokines compared to the M-AOM/DSS group at week 2 (Fig. 3B). Moreover, decreased levels of IL-6 and TNF-α were observed in the M-AOM/DSS+estr group compared to the M-AOM/DSS group (both p < 0.05) (Fig. 3B). The proinflammatory enzyme levels and cytokine gene expression at week 2 were consistent with the results of DAI and microscopic damage index at week 2. These data suggest that NF-κB and NF-κB–related pro-inflammatory mediators are the molecular basis of the sex difference in colitis. These observations also suggest that estradiol exerts anti-inflammatory effects by suppressing NF-κB–related pro-inflammatory mediators. NF-κB–related pro-inflammatory mediators were consistently expressed at high levels in the M-AOM/DSS-group (Fig. 3C-F).
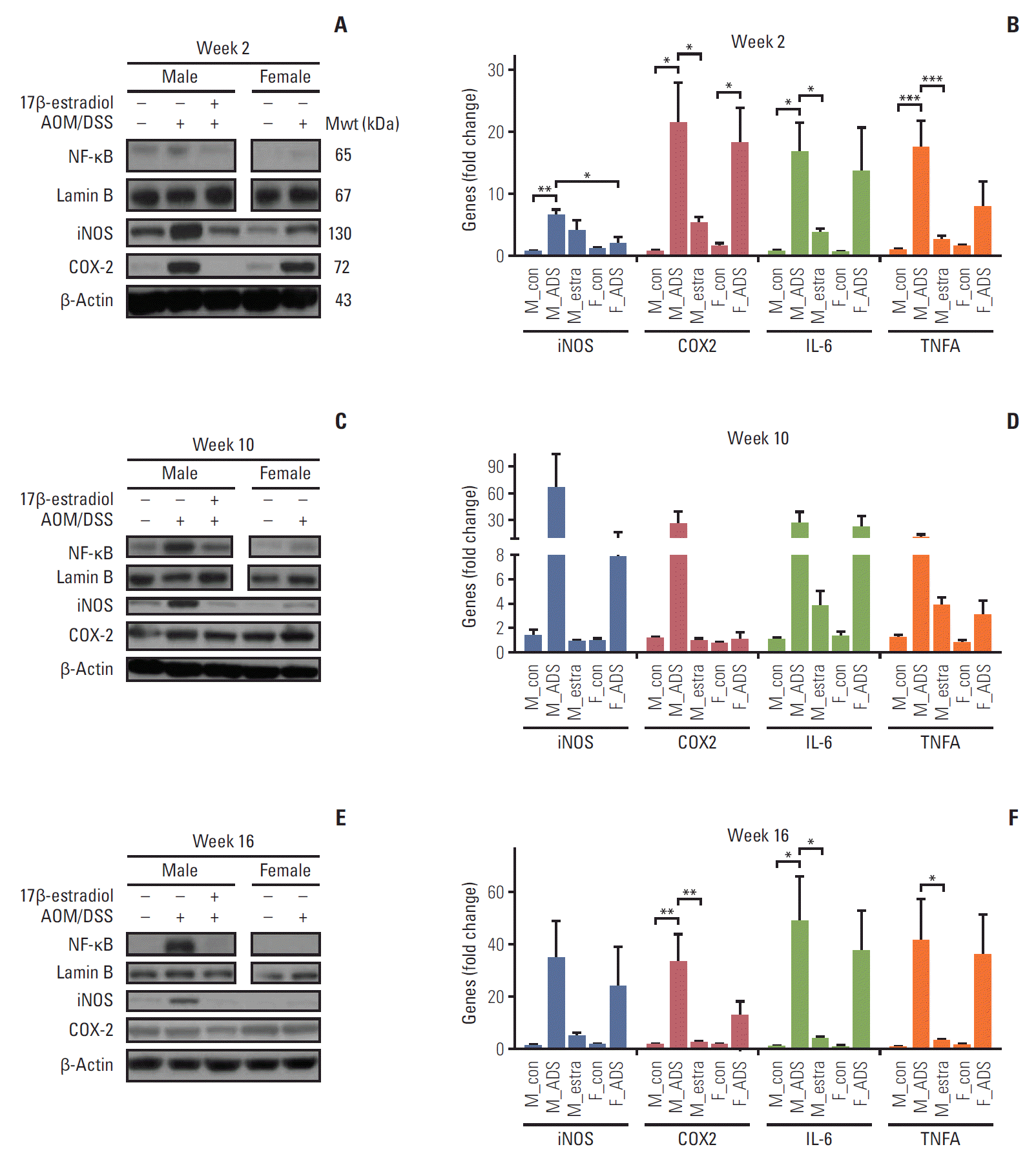
Fig. 3.
Protein and mRNA expression levels of nuclear factor κB (NF-κB) and its related pro-inflammatory factors in colonic tissues at weeks 2 (A, B), 10 (C, D), and 16 (E, F). Western blot analysis of NF-κB, inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX2) at weeks 2 (A), 10 (C), and 16 (E). mRNA expression levels of iNOS, COX2, interleukin 6 (IL-6), and TNFA, determined with real-time polymerase chain reaction, at weeks 2 (B), 10 (D), and 16 (F). *p < 0.05, **p < 0.01, and ***p < 0.001. M, male; F, female; AOM, azoxymethane; DSS, dextran sulfate sodium; estra, estradiol.
![]()
4. Effect of estradiol during colitis and cancer progression in terms of Nrf2
Since Nrf2 directly downregulates NF-κB expression and activity [13], we next investigated Nrf2 and its related antioxidant enzyme activation.
The IHC analysis of Nrf2 showed significant increase of Nrf2 by estradiol at week 2 (p < 0.05) (Fig. 4A and B), and by AOM/DSS at weeks 10 and 16 (p < 0.05 at week 10, p < 0.01 at week 16) (Fig. 4E, F, I, and J). The proportion of Nrf2-immunostained cells in crypts was significantly higher in females than in males on weeks 10 and 16 (all p < 0.05) (Fig. 4E, F, I, and J). At week 16, the Nrf2-immunostained cells were decreased by estradiol (p < 0.05).
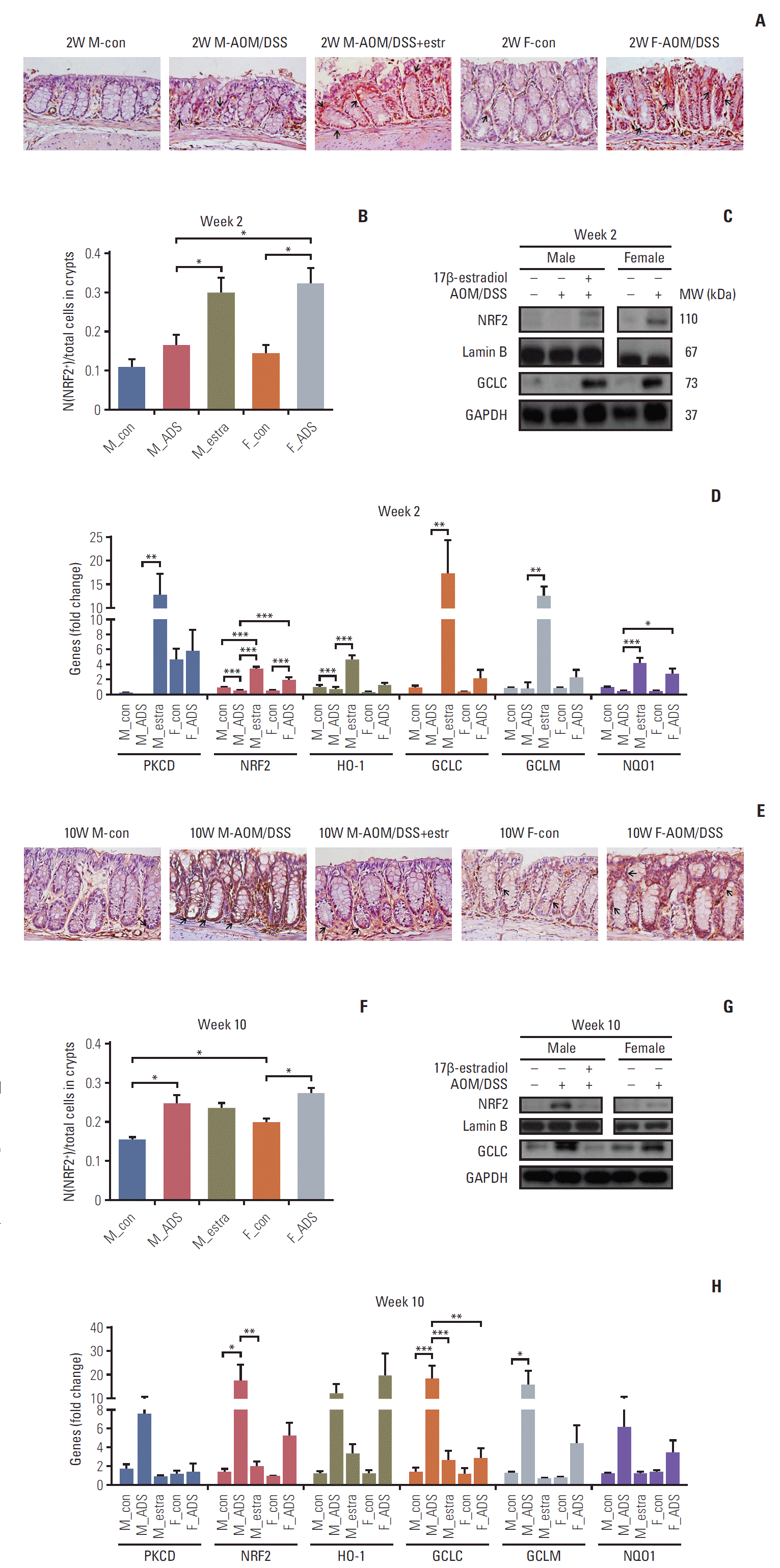
Fig. 4.
Expression levels of nuclear factor erythroid 2-related factor 2 (NRF2) and its related anti-oxidant enzymes in colonic tissues at weeks 2 (A-D), 10 (E-H), and 16 (I-L). Photomicrography of NRF2 immunostain of distal mouse colon at weeks 2 (A), 10 (E), and 16 (I). Arrows indicate the NRF2-immunoreactive cells (×400). Analysis of NRF2 immunohistochemistry in distal colonic tissues at week 2 (B), 10 (F), and 16 (J). Western blot analysis of NRF2 and glutamate-cysteine ligase catalytic subunit (GCLC) at weeks 2 (C), 10 (G), and 16 (K). mRNA expression levels of PKCD, NRF2, HO-1, GCLC, GCLM, and NQO-1, determined with real-time polymerase chain reaction, at weeks 2 (D), 10 (H), and 16 (L). *p < 0.05, **p < 0.01 and ***p < 0.001. M, male; F, female; AOM, azoxymethane; DSS, dextran sulfate sodium; ADS, AOM/DSS; estra, estradiol; MW, molecular weight; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
![]()
The F-AOM/DSS group showed higher expression of Nrf2 compared to the M-AOM/DSS group in terms of the levels of protein (Fig. 4C) and mRNA (Fig. 4D) at week 2. Nuclear translocation and mRNA expression of Nrf2 were increased in the M-AOM/DSS+estr group compared to the M-con and M-AOM/DSS groups at week 2 (p < 0.001) (Fig. 4C and D). The protein and mRNA levels of PKCδ, which positively regulate Nrf2 [8,21], also increased in the M-AOM/DSS+estr group compared to the M-AOM/DSS group (all p < 0.05) (Fig. 4D and S3 Fig. of the SI). The F-AOM/DSS and F-con groups also showed higher mRNA levels of PKCδ compared to the M-AOM/DSS and M-con groups (all p < 0.05) (Fig. 4D). In addition, Gα13 strengthens ERα activity [22], and estradiol is closely related to the activities of PKCδ [11] and Nrf2 [7]. Another G protein, Gα12, regulates NF-κB in an estradiol-independent pathway [23]. When we measured the Gα13 and Gα12 by Western blot analysis, only Gα13 was significantly increased in the M-AOM/DSS+estr group compared to the M-AOM/DSS group (p < 0.001) (S3 Fig.). Gα13 expression of the F-AOM/DSS group was significantly higher than the M-AOM/DSS (p < 0.001) (S3 Fig.). Taken together, these data strongly suggest that activation of Nrf2 by exogenous and endogenous estradiol is closely correlated to the activation of Gα13-PKCδ signaling pathway during DSS-induced inflammation stage at week 2.
Nrf2 activation also resulted in up-regulation of anti-oxidant gene expression at week 2. Glutamate-cysteine ligase catalytic subunit (GCLC) is an antioxidant enzyme regulated by Nrf2. GCLC was increased in the F-AOM/DSS and M-AOM/DSS+estr groups compared to control mice and the M-AOM/DSS group at week 2 (Fig. 4C). This observation is consistent with the increased mRNA expression of other antioxidant enzymes (i.e., HO-1, GCLM, and NQO-1) in the F-AOM/DSS and M-AOM/DSS+estr groups compared to control mice and the M-AOM/DSS group at week 2 (all p < 0.05 for M-AOM/DSS+estr group vs. M-AOM/DSS group, p < 0.001 for NQO-1 between F-AOM/DSS group and M-AOM/DSS group) (Fig. 4D). These data suggest that both endogenous and exogenous estradiol relieve DSS-induced colitis by promoting anti-oxidant gene expression through Nrf2 activation.
We investigated Nrf2 expression levels by Western blot analysis at weeks 10 and 16. At these times, CRC had developed in the AOM/DSS model. In contrast to week 2, both protein and mRNA levels of Nrf2 were higher in the M-AOM/DSS group compared to the M-AOM/DSS+estr group at weeks 10 and 16 (p < 0.01 at week 10) (Fig. 4E-L). Consistent with Nrf2, GCLC and other anti-oxidant enzymes were highly expressed in the M-AOM/DSS-group at weeks 10 and 16 (Fig. 4H and L). These findings suggest that, after tumor initiation, Nrf2 expression might overly induce antioxidant enzymes that could somehow result in a tumor microenvironment that was favorable to tumor progression. This explanation was supported by the cancer developmental factor analysis at the F-AOM/DSS group. That is, 25% of weeks 10 and 16 (both n=12) showed CRC development; they were divided into the cancer group (both n=4) and non-cancer group (both n=8) (S4 Fig.). The F-AOM/DSS cancer group showed significantly higher levels of HO-1 and PKCδ than the F-AOM/DSS non-cancer group at week 16 (p < 0.05 for PKCδ) (S4 Fig.). Other factors, such as NQO1, GCLM, iNOS, and Nrf2, were elevated in the F-AOM/DSS cancer group compared to the F-AOM/DSS non-cancer group, but did not reach statistical significance (S4 Fig.).
5. Effect of estradiol during colitis and cancer progression in terms of NLRP3
The close relationship of Nrf2 with the activating mechanism of NLRP3 inflammasome, which finally activates IL-1β and IL-18 [16], inspired the present measurement of protein and mRNA levels of the NLRP3 inflammasome and its related mediators by Western blot (Fig. 5A) and qRT-PCR analyses (Fig. 5B), to investigate its relationship with Nrf2 in AOM/DSS mice. Consistent with the Nrf2-mediated activation and up-regulation of anti-oxidant gene expression at week 2, the Western blot analysis clearly showed an increase of the NLRP3 inflammasome and its related phosphorylated caspase-1 and IL-1β in the F-AOM/DSS and M-AOM/DSS+estr groups compared to the M-AOM/DSS group at week 2 (p < 0.05 for NLRP3 and IL-1β between the M-AOM/DSS+estr and M-AOM/DSS groups) (Fig. 5A, S3 Fig.). The data for mRNA expression definitely supported the significant increase of IL-1β in the M-AOM/DSS+estr group compared to the M-AOM/DSS group, (p < 0.001) (Fig. 5B).
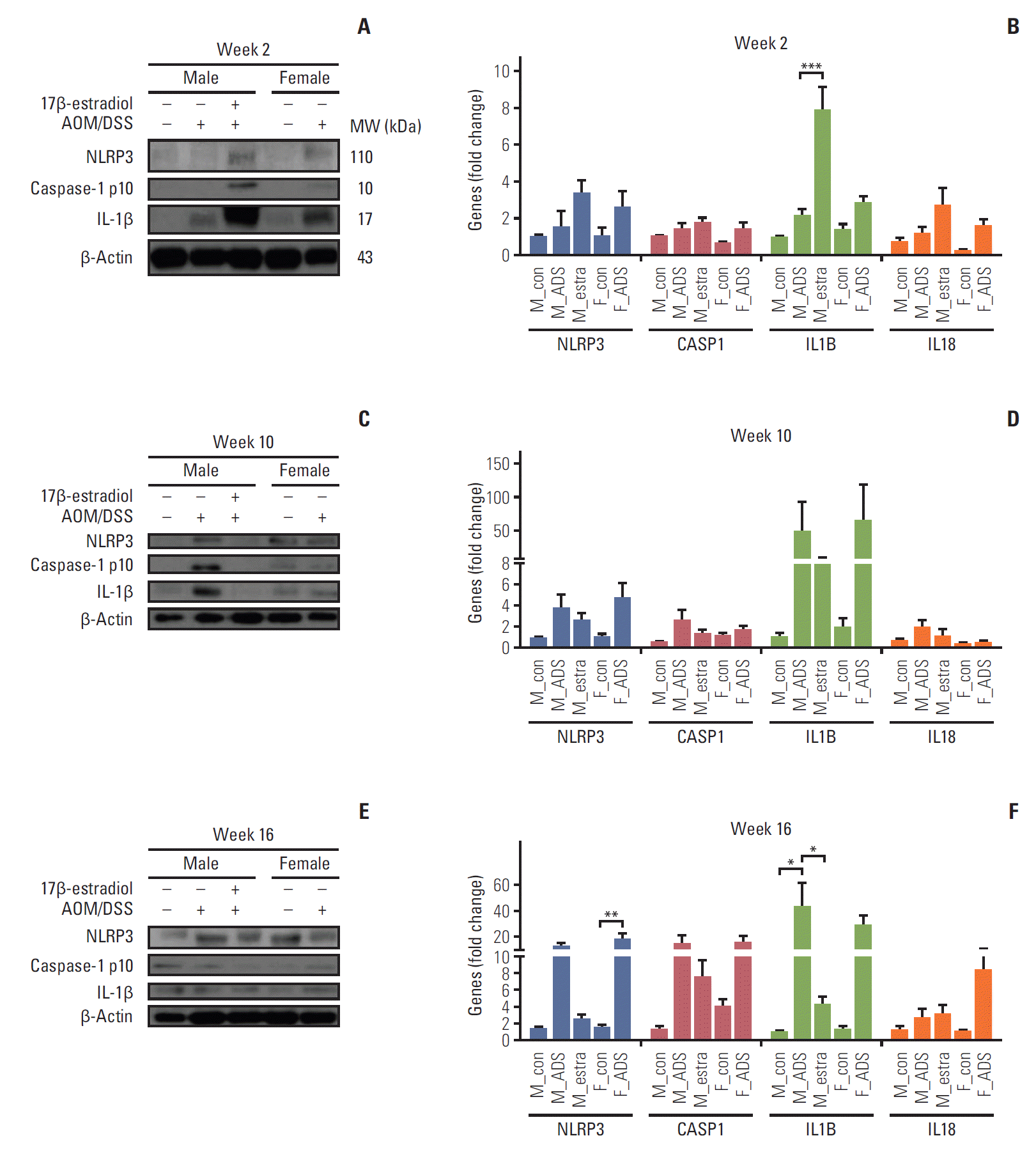
Fig. 5.
Protein and mRNA level analyses of Nod-like receptor protein 3 (NLRP3) inflammasome activation in colonic tissues at weeks 2 (A, B), 10 (C, D), and 16 (E, F). Western blot analysis of NLRP3, caspase-1 p10, and interleukin (IL)-1β at weeks 2 (A), 10 (C), and 16 (E). mRNA expression levels of NLRP3, CASP1, IL1B, and IL18, determined with real-time polymerase chain reaction, at weeks 2 (B), 10 (D), and 16 (F). *p < 0.05, **p < 0.01, and ***p < 0.001. M, male; F, female; AOM, azoxymethane; DSS, dextran sulfate sodium; MW, molecular weight; ADS, AOM/DSS; estra, estradiol.
![]()
Next, we examined the expression of NLRP3 inflammasome-related enzymes and mediators (NLRP3, caspase 1, IL-1β, and IL-18) at weeks 10 and 16 by Western blot analysis. Notably, the expressions of caspase-1 and IL-1β were elevated in the M-AOM/DSS group compared to the M-AOM/DSS+estr group at weeks 10 and 16 in both protein (Fig. 5C and E) and mRNA analyses (Fig. 5D and F). Similarly, the F-AOM/DSS cancer group also showed higher mRNA levels of NLRP3 inflammasome-related enzymes and mediators compared to the F-AOM/DSS non-cancer group (p < 0.05 for NLRP3 and caspase-1 at week 10, p < 0.05 for NLRP3 at week 16) (S4 Fig.). These data suggest that once a tumor is initiated, the NLRP3 inflammasome might promote tumor development, which is consistent with the Nrf2 data.
Discussion
After confirming the sex difference in CRC development by showing that the F-AOM/DSS group has significantly lower tumor multiplicity and incidence compared with the M-AOM/DSS group, we further investigated the underlying anti-cancer mechanism of estradiol. Our findings demonstrate a dual role of Nrf2 in modulating inflammation and carcinogenesis through the regulation of the NF-κB–mediated pro-inflammatory pathway, anti-oxidant enzymes, and NLRP3 inflammasome. In this research, we focused on week 2, which is the active DSS-induced inflammation stage [20], just after the completion of estradiol administration, and several weeks before the AOM/DSS-induced tumorigenesis. The severity of inflammation at week 2 was associated with tumor formation at weeks 10 and 16. Our study clearly demonstrates the importance of early inflammatory control by the administration of estradiol to AOM/DSS-treated male mice, to confirm the role of estradiol for CRC prevention. In addition, the M-AOM/DSS+estr group was compared with the F-AOM/DSS group to check any differences between exogenous and endogenous estradiol. This approach strongly supports the inhibitory effect of estradiol on inflammation and inflammation-induced tumorigenesis, and also notably uncovers its underlying mechanism of estradiol in three aspects: (1) NF-κB–related pro-inflammatory mediators, (2) Nrf2-related anti-oxidant enzymes, and (3) NLRP3 inflammasome.
The NF-κB signaling pathway is highly involved in inflammation and cancer development, especially in colitis-associated CRC [24]. In the present study, estradiol inhibited the NF-κB signaling pathway involving iNOS, COX-2, IL-6, and TNF-α during the DSS-induced inflammation stage of AOM/DSS-induced colon tumorigenesis. Furthermore, the concomitant increased expression of Gα13, PKCδ, and Nrf2 by estradiol administration in AOM/DSS-treated male mice during the DSS-induced inflammation stage supports the correlation between the suppression of NF-κB signaling and the estradiol-induced activation of Gα13/PKCδ/Nrf2 pathway. ERs inhibit the NF-κB pathway by the modulation of upstream signaling or the transcriptional activation of NF-κB in various cell lines [25]. Nrf2 inhibition of NF-κB activity is well known. Gα13 strengthens ERα activity [22], and estradiol regulates the activity of PKCδ [11], and Nrf2 [7]. To further support the estradiol-induced activation of Nrf2 pathway, we are performing experiments using Nrf2 Knockout mice.
It has been reported that anti-oxidant enzymes activated by Nrf2 have cancer preventive effects by eliminating reactive oxygen species, and facilitating the resolution of inflammation [26]. In the present study, the levels of Nrf2 and NQO-1 expression were different between the M-AOM/DSS and F-AOM/DSS groups, implicating the Nrf2-related antioxidant reaction as an underlying mechanism of the protective effect of estradiol. The high expression levels of Nrf2 and its related anti-oxidant enzyme in the M-AOM/DSS+estr group further strengthen this suggestion.
There was an increase of the NLRP3 inflammasome and its effector cytokines (IL-1β and IL-18) simultaneously with increased Nrf2 on the DSS-induced inflammation stage. Considering the importance of Nrf2 in NLRP3 inflammasome activation [15], Nrf2 activation might lead to immune modulation through caspase-1 related activities, such as pyroptosis [16]. Based on the knowledge that NLRP3 inflammasome activation induces pyroptosis [16], we hypothesize that Nrf2/NLRP3 inflammasome/IL-1β–mediated pyroptosis triggers the elimination of precancerous cells during inflammation, and further prevents carcinogenesis in the presence of estradiol (Fig. 5A). To prove this hypothesis, further investigations are required.
Since estradiol administration completely inhibited inflammation in AOM/DSS-treated male mice resulting in the near-complete prevention of CRC, we expected that AOM/DSS-induced inflammation at week 2 would be mild in female mice. There was a significant difference of DAI at week 2 between male and female mice, but not such significant differences in the severity of inflammation reflected in COX-2. The inflammation represented by DAI cannot be fully explained by a few inflammatory mediators, such as COX-2. The strong effect of administered estradiol on preventing AOM/DSS-induced inflammation and colon tumorigenesis might be due to the higher concentration of intraperitoneal-administered estradiol; the injection concentration was 10 mg/kg, compared to the concentration of endogenous estradiol of 4 pg/mL in female mice [27]. Furthermore, the endogenous estradiol level in mice sacrificed at week 2 might be lower than that of fully developed female mice. Also, sex difference in stress susceptibility may affect levels of some pro-inflammatory cytokines (e.g., IL-1β) [28], implying that endogenous estradiol has more complex action than exogenously injected estradiol. However, in terms of cancer prevention, as shown in S4 Fig. that compares the F-AOM/DSS non-cancer group with the F-AOM/DSS cancer group, endogenous estradiol seems to have a similar effect and mechanism, including PKCδ and inflammasome with exogenous estradiol. Different methods of estradiol treatment, various estradiol concentrations, and blood estradiol level monitoring can be considered to provide more physiologic conditions. To further investigate the effect of endogenous estradiol and its underlying mechanism, experiments using the ovariectomized female mice are underway.
After tumors developed (weeks 10 and 16), we observed the interesting finding that Nrf2 signaling was significantly up-regulated in the M-AOM/DSS group, suggesting that Nrf2 and anti-oxidant enzymes might play a role in promoting tumor progression (Fig. 5B). Several studies reported the possibility of the dual role of Nrf2 in tumor prevention and progression [29]. Satoh et al. [29] showed that Nrf2 activation in cancer cells enhances tumor malignancy, while Keap1-knockdown mice having high expression of Nrf2 are more resistant to urethane-induced carcinogenesis. Similar to Nrf2, we found two facets of NLRP3 inflammasome. Although NLRP3 inflammasome is a well-established target of NF-κB, its expression is inversely related to NF-κB expression at week 2. This indicates that it might have different roles from NF-κB, such as inducing pyroptosis in the DSS-induced inflammation stage. In contrast, it has been reported that once tumor formation is initiated, NLRP3 inflammasomeinduced IL-1β and IL-18 modulate immunity in the tumor microenvironment, and promote cancer progression [30]. A significant increment of NLRP3 and caspase-1 expression in the F-AOM/DSS cancer group compared to the F-AOM/DSS non-cancer group in the present study also supports tumor promotion by the NLRP3 inflammasome.
The collective present and prior [8,16,22] data support the proposal that the regulatory mechanism of estradiol in colitis-associated CRC depends on sex, and the timing of DSS-induced inflammation and carcinogenesis (Fig. 6). At the peak of inflammation at week 2, estradiol appears to induce inflammasome activation through Gα13 protein subunits. Gα12 and Gα13 have potentiated estradiol-bound ERα activity [22]. However, despite the functional overlap between Gα12 and Gα13, only Gα13 regulates NRF2 via PKCδ [8]. NRF2 mediates inflammasome activation through the transcription of as-yet unknown genes. NLRP3 inflammasome activation induces pyroptosis to eliminate precancerous cells [16]. NRF2 inhibits NF-κB, which is activated by inflammatory activators through Toll-like receptor signaling and reactive oxygen species. Ultimately, estradiol prevents carcinogenesis, whereas in the absence of estradiol, a cancer inducing microenvironment is created through NF-κB activation. When precancerous cells are not completely eliminated, cancer progresses through both the Gα12 and Gα13 protein subunits. Gα12 regulates the NF-κB mediated signaling pathway [8,23]. Gα13 regulates NRF2 via PKCδ [8]. NRF2 promotes tumor progression by the activation of anti-oxidant enzymes and NLRP3 inflammasome [15]. Ultimately, NF-κB and NRF2 signaling pathways accelerate carcinogenesis.
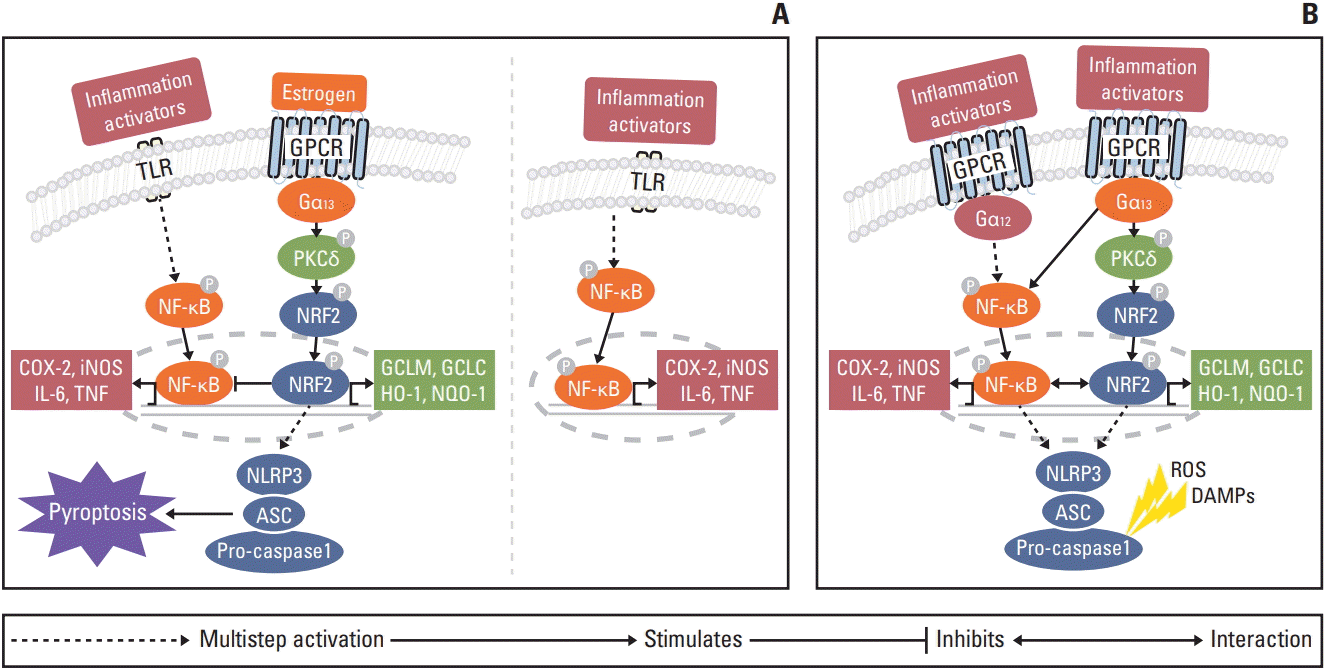
Fig. 6.
Proposed regulatory mechanism of estrogen in colitis-associated colorectal cancer at week 2 (A) and at weeks 10 and 16 (B). (A) Estrogen induces inflammasome activation through Gα13 protein subunits. Gα12 and Gα13 have potentiated estrogen-bound estrogen receptor α activity. However, despite the functional overlap between Gα12 and Gα13, only Gα13 regulates nuclear factor erythroid 2-related factor 2 (Nrf2) via protein kinase Cδ (PKCδ). Nrf2 mediates inflammasome activation through the transcription of as-yet unknown genes. Nod-like receptor protein 3 (NLRP3) inflammasome activation induces pyroptosis to eliminate precancerous cells. After eliminating precancerous cells, Nrf2 inhibits nuclear factor κB (NF-κB) and reactive oxygen species through the anti-oxidant enzymes. Ultimately, estrogen prevents carcinogenesis (left panel). In contrast, in the absence of estrogen, inflammation provides a cancer microenvironment through activation of the NF-κB pathway (right panel). (B) After unsuccessful elimination of precancerous cells, inflammation progresses to cancer at weeks 10 and 16. Gα12 and Gα13 regulate NF-κB and Nrf2 via PKCδ-mediated signaling pathway, respectively. Nrf2 promotes tumor progression by activation of anti-oxidant enzymes and NLRP3 inflammasome. Ultimately, NF-κB and Nrf2 signaling pathway accelerate carcinogenesis. COX-2, cyclooxygenase 2; DAMP, damage-associated molecular pattern; GPCR, G protein coupled receptor; IL, interleukin; iNOS inducible nitric oxide synthase; ROS, reactive oxygen species; TLR, Toll-like receptor; TNF, tumor necrosis factor.
![]()
In conclusion, our study shows estradiol administration in AOM/DSS-treated male mice attenuates inflammation, and increases Nrf2 in the DSS-induced inflammation stage. Moreover, inhibition of the NF-κB-related pathway and activation of Nrf2-related anti-oxidant enzymes and the NLRP3 inflammasome pathway indicate possible underlying mechanisms. This study finally suggests that Nrf2 and the NLRP3 inflammasome play a dual role, with a preventive effect on tumor development, but promotion of tumor progression, once a tumor is initiated.
 XML Download
XML Download