

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC), especially rectal cancer (RC), is one of the most common malignancies in China [1]. The presurgical radiotherapy (RT) is now a necessary constituent of the standard treating mode for locally advanced RC [2]. The tumor regression grade (TRG) after RT has been proven as an independent factor predicting the long-term survival of the patients [3]. However, even after standard chemoradiotherapy (CRT), only 21.3% of the patients attained pathologic complete remission (pCR). And the good responders occupy merely 31.2% of the cases [4]. To achieve success of RT and improve prognosis, cellular resistance to ionizing radiation (IR) remains the primary barrier to overcome.
DNA damage repair (DDR) is acknowledged as the most classical mechanism of cellular radioresistance [5]. And recent studies revealed that tumor microenvironment (TME) also played important roles in radiosensitivity modulation [6]. Chemokine receptors, a kind of G protein–coupled transmembrane molecules regulating organogenesis and inflammation under physiological state, are now known as bridges through which the TME impacts many biological procedures within the tumor cells [7]. The C-C motif chemokine receptor 6 (CCR6) is the most-studied chemokine receptor in CRC. It is overexpressed abnormally in CRC cells and the surrounding tumor-associated immune cells. And a negative association have been reported between CCR6 expression and the patients’ clinical outcome [8,9]. The CCR6 has also been demonstrated to promote oncogenesis, progression, and metastasis of CRC [9,10]. Yet, there is no evidence on the functions of CCR6 in RC radioresistance until now. Since the downstream signaling molecules of CCR6, the Akt and the ERK, could regulate DDR through enhancing the stability of the direct participants, such as DNA-PK and Rad51 [11-14], we hypothesized that CCR6 might influence radiosensitivity of RC, and be a new target for radiosensitization therapy. In this study, we determined the relationship between CCR6 expression and RC radioresistance, through patients and cell lines exhibiting different response to IR. The modulating ability of CCR6 on radiosensitivity was further confirmed by CCR6 knockdown.
Materials and Methods
1. Tissue specimens
The pretreatment tumor tissue of the patients who had pathologically diagnosed RC and received CRT was from the tumor bank of the Sun Yat-sen University Cancer Center (SYSUCC). The paraffin-embedded specimens were for immunohistochemical (IHC) staining, and the specimens contained in RNAlater (Thermo Fisher Scientific, Waltham, MA) were for RNA sequencing (RNA-seq).
2. Evaluation of TRG
The postoperative pathology of each patient treated with radical resection was assessed by an experienced attending pathologist. The TRG was determined according to the Mandard standard [4], in which pCR was defined as TRG 1.
3. RNA-seq
Among the patients receiving surgery between May 1, 2013 and August 31, 2013, six cases were randomly selected from those exhibiting pCR in the postsurgical pathology. And another six cases were randomly selected from those exhibiting non-pCR. RNA-seq was performed for these 12 patients, using the HiSeq 4000 system (Illumina, San Diego, CA). The pathoclinical profiles of the patients were shown in S1 Table (see Supplementary Materials).
The expression level was represented with fragments per kilobase million (FPKM) [15]. The fold change (FC) of each gene was calculated through dividing the average FPKM of the non-pCR group by that of the pCR group. The difference in the average FPKM between the two groups was tested by a student’s t test. Hierarchical clustering analysis based on Euclidean distance was performed in genes with a p-value of < 0.01, through MultiExperiment Viewer 4.9.0 (available at http://tm4.org). And, pathway enrichment analysis was also made in those genes through Metascape [16].
4. IHC analysis
IHC analysis was performed in the consecutive patients who received surgery between November 1, 2015 and September 30, 2017. Before staining, all the paraffin-embedded specimens were deparaffinized with xylenes, rehydrated with graded ethanol to distilled water, and submerged in EDTA antigen retrieval buffer (1 mmol/L, pH 8.0) and microwaved to retrieve antigens. IHC staining was then performed using the Dako REAL Envision system, Peroxidase/DAB, Rabbit/Mouse (Dako, Carpinteria, CA), following the manufacturer’s recommended protocols. After treatment with 0.3% hydrogen peroxide for 15 minutes and normal goat serum for 30 minutes, the specimens were incubated with a CCR6 rabbit polyclonal antibody (1:100, ab109703, Abcam, Cambridge, UK) overnight at 4°C. The sections were then incubated with a peroxidase-conjugated Dako REAL EnVision/HRP, Rabbit/Mouse (ENV) reagent (solution A) for 30 minutes. Finally, the visualization was done by incubating the sections in DAKO REAL DAB+ Chromogen (solution C) for 10 minutes. The slides were washed with phosphate buffered solution (PBS) between the staining steps.
The expression of CCR6 was evaluated by another experienced attending pathologist who was blinded to the TRG of the patients. The entire tissue section was observed to assign scores of intensity and extent. The intensity scores included as 0 (no staining), 1 (light yellow), 2 (yellow brown), and 3 (brown). The extent scores were decided according to the percentages of the positive cells in the whole carcinoma area, included 0 (0%), 1 (1-25%), 2 (26-50%), 3 (51-75%), and 4 (76-100%). The final staining score (0 to 12) was the product of the intensity and extent scores. Tumors with a staining score of < 4 were defined as low expression, and those with a score ≥ 4 were defined as high expression (S2 Fig.).
The median IHC staining scores of the resistant group (patients with TRG 3-5) and the sensitive group (patients with TRG 1-2) were compared through a Mann-Whitney U test. And proportions of the cases with high CCR6 expression in these two groups were also compared, through a chi-square test.
5. Cell culture
The CRC cell lines used in this study included LoVo, sw480 and sw620, which were purchased from the American Type Culture Collection (Manassas, VA) and maintained in the SYSUCC [17]. These cells were all cultured in Roswell Park Memorial Institute 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), penicillin (100 units/mL), and streptomycin (100 units/mL). An incubator was used to create a humidified atmosphere of 5% CO2 and a constant temperature of 37°C for cell culture.
6. RNA interference
To knockdown the CCR6 expression, transient transfection of small interfering RNA (siRNA) was performed, using the Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific). The siRNA duplexes were synthesized by the Genechem Co. (Shanghai, China), and consisted of 5′-GGUCUAUGACAGACGUCUAUCdTdT-3′ and 5′-UAGACGUCUGUCAUAGACCUGdTdT-3′ as the sense and antisense sequences, respectively. The AllStars Negative Control siRNA (Qiagen, Hilden, Germany) was used as the negative control. The cells were harvested 24 hours after transfection for the subsequent experiments.
7. Irradiation and clonogenic survival assay
The cells were trypsinized and seeded into 6-well plates. Different cell quantities were for different doses of irradiation (100, 200, 103, and 104 cells were for 0, 2, 4, and 6 Gy, respectively).The irradiation was performed by a R2000 X-ray irradiator (Rad Source Technologies, Suwanee, GA), with a dose rate of 1.1 Gy/min, a voltage of 160 kV, a current of 25 mA, and 0.3-mm copper filters. The cells were then cultured as we described above for 14 days, fixed by 4% paraformaldehyde, and stained with Giemsa. Colonies with more than 50 cells were scored as survivors. The plating efficiency (PE) was calculated by dividing the number of survivors by the number of plated cells at 0 Gy. Surviving fraction (SF) at each dose was determined as the number of survivors divided by the product of the PE and the number of plated cells at the corresponding dose. Sensitization enhancement ratio (SER) was calculated by division of SF at 2 Gy (SF2). Clonogenic survival curves were fitted to the linear quadratic model, SF=exp[-(αD+βD2)], and compared through the extra-sum-of-squares F test.
8. Western blot analysis
The expression level of CCR6 protein in CRC cells, including those transfected with siRNA, was examined by western blot analysis. The procedure of western blot referred to a report of Cerda et al. [18] prior to our study. After lyses with the RIPA buffer, the total protein in the cells were collected at 4°C and quantified by a Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Protein of 50 μg was mixed with sample buffer, which consisted of 250 mmol/L Tris-HCl (pH 6.8), 8% sodium dodecyl sulfate (SDS), 0.04% bromophenol blue, 40% glycerol, and 20% β-mercaptoethanol. The mixture was heated for 5 minutes at 95°C, resolved by SDS polyacrylamide gel electrophoresis (the concentration of polyacrylamide gel was 10%), and transferred to nitrocellulose membranes (Thermo Fisher Scientific) by electroblotting. The membranes were blotted with 5% skimmed milk, washed with Tris-buffered saline Tween, and incubated with the primary anti-CCR6 rabbit polyclonal antibody (1:1,000, ab109-703, Abcam) overnight at 4°C. After washing, the membranes were incubated with anti-rabbit horseradish peroxidase conjugated IgG (1:2,000, Santa Cruz Biotechnology, Santa Cruz, CA) for 1 hour. Fluorescence detection was then performed through the enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. The integrated option density (IOD) of a specific band was measured by Image-pro Plus 6.0 (Media Cybernetics, Rockville, MD). The CCR6 expression level in each cell line was represented with the IOD of the CCR6 band divided by the IOD of the GAPDH band. The FC was calculated by dividing the CCR6 expression level in a specific cell line by that in the reference cell line.
9. Reverse transcription-polymerase chain reaction
The expression level of CCR6 mRNA in CRC cells was examined by reverse transcription-polymerase chain reaction (RT-PCR) analysis. Total RNA from different CRC cell lines were extracted with Trizol reagent (Thermo Fisher Scientific). Then the first-strand cDNA was synthesized with 1 μg of total RNA, through the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). Realtime RT-PCR was performed in triplicate for each sample, using the Absolute qPCR SYBR Green Mixes (Thermo Fisher Scientific). PCR reaction and data collection were done through the ABI PRISM7900HT sequence detection system (Thermo Fisher Scientific). GAPDH was used as the endogenous control for normalization. The primer sequences for CCR6 were 5′-ACCGCAGATAACGACAATGC-3′ (sense) and 5′-CATGAGCACGTTAAGTCCCG-3′ (anti-sense), which were designed by the Primer3 4.1.0 [19]. And the primer sequences for GAPDH were 5′-CTCCTCCTGTTCGACAGTCAGC-3′(sense) and 5′-CCCAATACGACCAAATCCGTT-3′ (antisense) [20].
10. Immunofluorescence analysis
Immunofluorescence (IF) staining countering phosphorylated histone H2AX (γH2AX) was made to evaluate the DDR abilities of CRC cells [20]. First, the cells were seeded on coverglasses in 24-well plates (5×104 cells per well), cultured for 24 hours, and irradiated with an X-ray of 2 Gy. At 30 minutes and 24 hours after irradiation, the cells were fixed (4% paraformaldehyde, 15 minutes) and permeabilized (0.25% Triton-X 100 in PBS, 15 minutes) at 4°C. Blocking was carried out by adding 5% bovine serum albumin in PBS for 30 minutes. Next, the cells were incubated with an anti-γH2AX (Ser139) rabbit polyclonal antibody (1:1,000, ab2893, Abcam) for 2 hours, followed by Alexa 488-conjugated goat anti-rabbit IgG (1:500, ab150077, Abcam) for 1 hour. The nuclei were counter stained with 1 μg/mL DAPI solution (Thermo Fisher Scientific). Finally, coverslips were mounted with Vectashield (Vector Laboratories, Peterborough, UK). Images were then taken through an Olympus FV100 confocal imaging system (Olympus Co., Tokyo, Japan). At each time point, IF staining was conducted in triplicate for each cell line. Difference in average number of the γH2AX-positive cells between any two cell lines at a defined time point were compared by a student’s t test.
Results
1. CCR6 expression was inversely correlated with radiosensitivity of RC patients
To determine how CCR6 influenced the treatment effects of RT, we first assessed the expression level of CCR6 mRNA through RNA-seq in RC patients exhibiting different response to RT. Among the 34,316 genes sequenced, there were totally 22,978 genes presenting expression. Of those, differential expression (p < 0.05) was seen in 768 genes, in which 116 genes exhibited obviously differential expression (p < 0.01), including the CCR6 gene. The volcano plot and the clustering heatmap of these 116 genes were shown as Fig. 1A and B, respectively. The average FKPM of CCR6 gene in the non-pCR group was greater than that in the pCR group (FC, 2.11; p=0.004). Hence, the CCR6 mRNA was expressed more highly in the non-pCR patients than in the extremely good (pCR) responders. Additionally, the chemokine-mediated signaling pathway in which CCR6 took part was one of the top 5 enriched pathways (Fig. 1C).
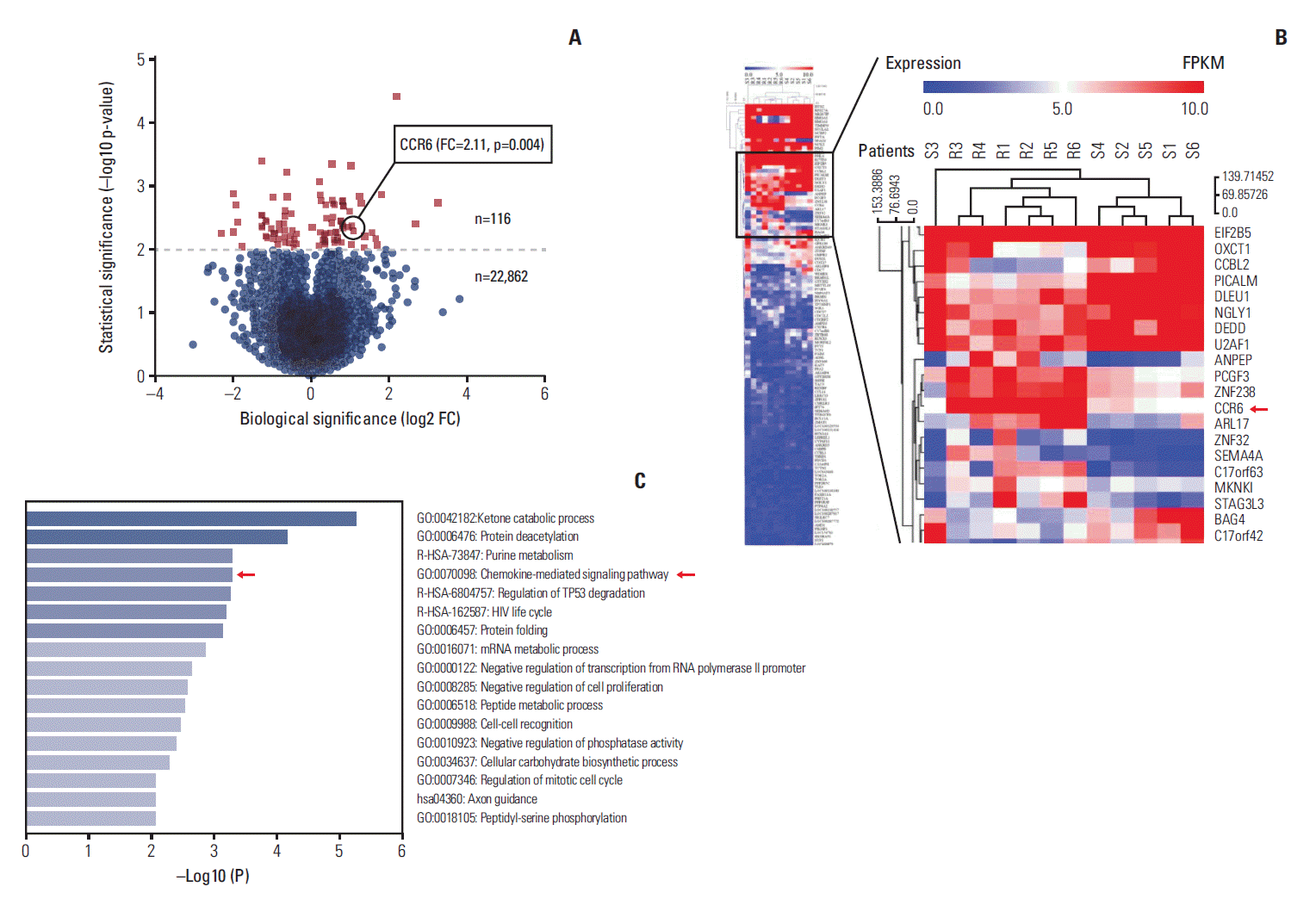
Fig. 1.
Profiles of RNA sequencing data. (A) Volcano plot of the 22,978 expressed genes. (B) Clustering heatmap of the 116 genes exhibiting obviously differential expression (p < 0.001). The C-C motif chemokine receptor 6 (CCR6) expression level was higher in the non-pathologic complete remission patients (R1-R6) than in the pCR patients (S1-S6). The fold change (FC) was 2.11 (p=0.004). (C) Heatmap of the pathway enrichment analysis. The top 5 enriched pathways were ketone catabolic process, protein deacetylation, purine metabolism, chemokine-mediated signaling pathway, and regulation of TP53 degradation.
![]()
To confirm the results of RNA-seq, we performed evaluation on expression level of CCR6 protein in an enlarged sample of patients. There were a total of 95 patients undergoing IHC analysis, including 55 cases in the resistant (TRG 3-5) group and 40 patients in the sensitive (TRG 1-2) group. Except CCR6 expression, the pathoclinical profiles of the patients were balanced between the two groups (S3 Table). The median score of the resistant group was greater than that of the sensitive group (8 vs. 3, p=0.005) (Fig. 2A). And high-level CCR6 expression was more common in the resistant group than in the sensitive group (76.3% vs. 37.5%, p < 0.001) (Fig. 2B). The results indicated that CCR6 protein was expressed more highly in the bad responders than in the good responders.
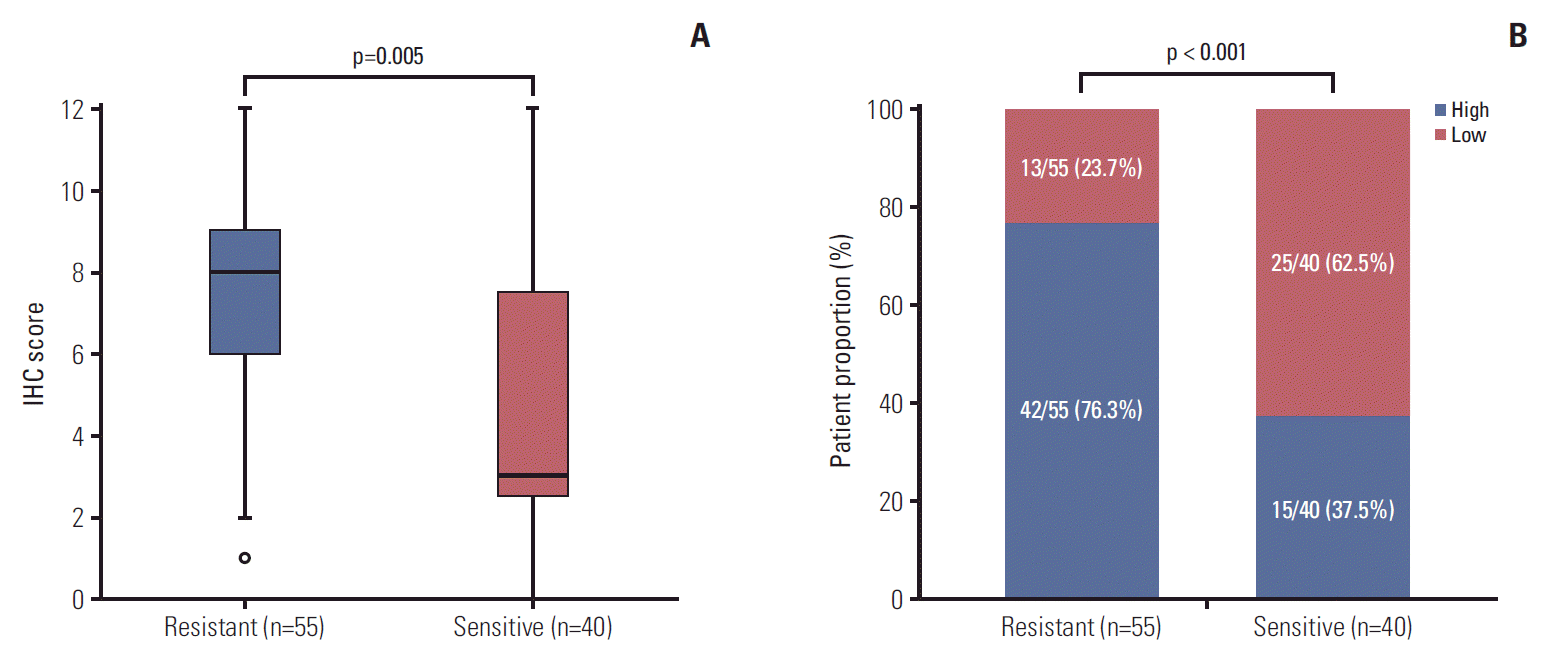
Fig. 2.
Results of immunohistochemical (IHC) analysis. (A) Median IHC score of the resistant group was greater than that of the sensitive group (8 vs. 3, p=0.005). (B) Patients with high-level C-C motif chemokine receptor 6 expression were more common in the resistant group than in the sensitive group (76.3% vs. 37.5%, p < 0.001).
![]()
Taking these data together, CCR6 expression was inversely correlated with RT effects of the RC patients.
2. CRC cells with high CCR6 expression was resistant to irradiation
To know the exact role of CCR6 in radioresistance of RC, CCR6 expression level was tested in different CRC cell lines. The results of western blot and RT-PCR analyses were showed as Fig. 3A and B, respectively. The sw480 and LoVo cells presented a relatively high level of CCR6 expression. Contrarily, there was a relatively low level of CCR6 expression in the sw620 cells. The clonogenic survival assay indicated that CRC cell lines were arranged in descending order of radioresistance as following: LoVo≈sw480 > sw620 (Fig. 3C). The SF2 of the three cell lines were 24.6%, 22.8%, and 17.8%, respectively (Table 1). Thus, there was an up-regulation of CCR6 expression in the cell lines relatively resistant to IR. It indicated that CCR6 might participate in regulation of RC radiosensitivity.
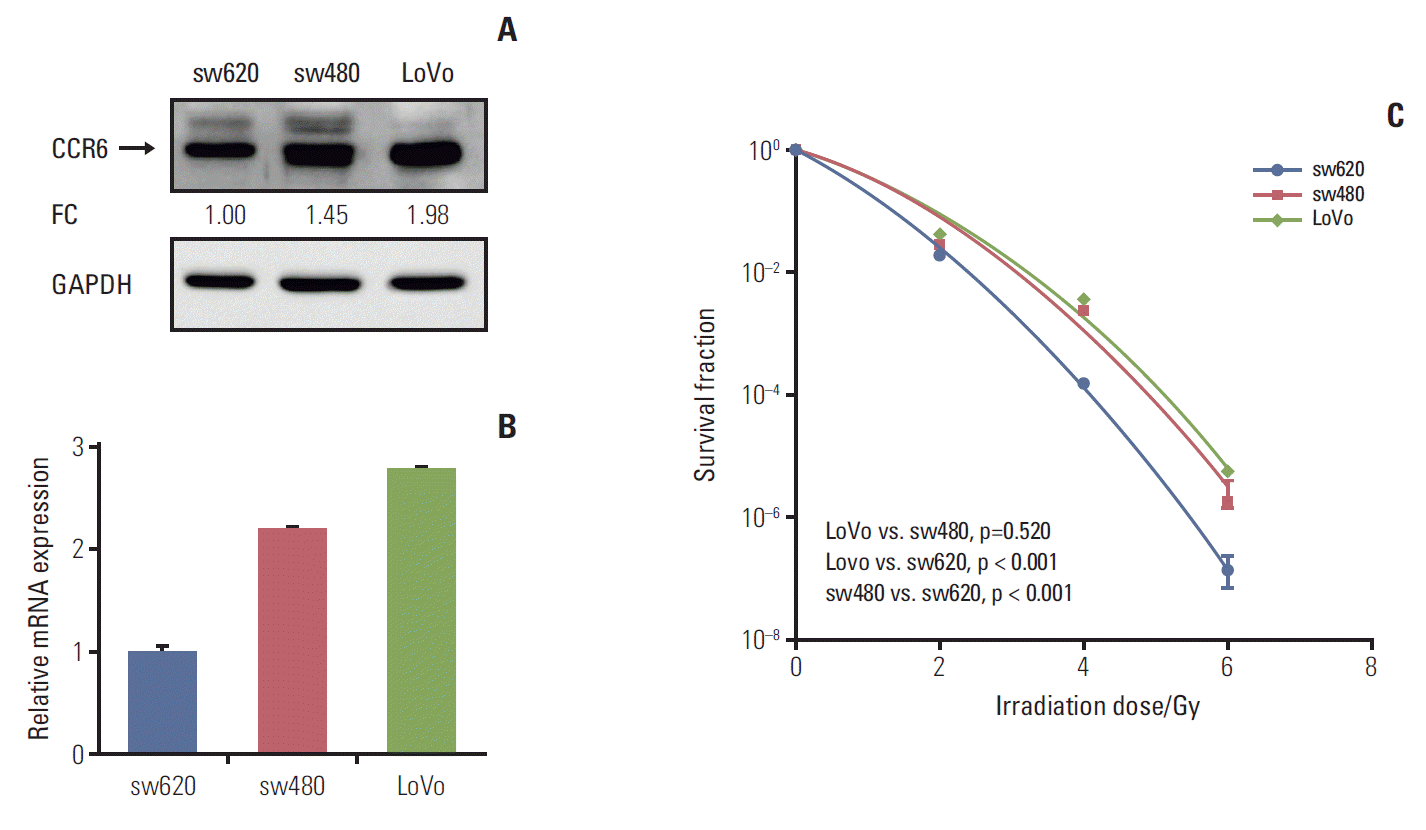
Fig. 3.
Colorectal cancer (CRC) cell lines with high C-C motif chemokine receptor 6 (CCR6) expression were resistant to ionizing radiation. (A) Western blot analysis on CCR6 expression in the sw620, the sw480 and the LoVo cell lines. The reference cell line to calculate the fold change (FC) was the sw620 cell line. (B) Real-time polymerase chain reaction analysis on CCR6 expression in the CRC cell lines described in panel A. (C) Postirradiation survival curves of the CRC cell lines described in panel A. The sw480 and LoVo cells which presented higher CCR6 expression appeared to be more resistant to ionizing radiation. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
![]()
3. CCR6 knockdown sensitized CRC cells to irradiation
In vitro silencing targeting CCR6 was performed in the LoVo cells which expressed relatively high level of CCR6 and had the greatest SF2, to validate the impact of CCR6 on radiosensitivity. The silencing efficiency was confirmed by western blot and RT-PCR analyses (Fig. 4A and B). The clonogenic survival assay showed that the siRNA-transfected LoVo cells (LoVo-si) were more sensitive to IR, compared with the LoVo cells. The SER was 1.738 (p < 0.001) (Fig. 4C). No radiosensitizing effect was seen in the LoVo cells transfected with negative control (LoVo-nc). The results of RNA interference further supported the ability of CCR6 in modulating the sensitivity of RC to IR.
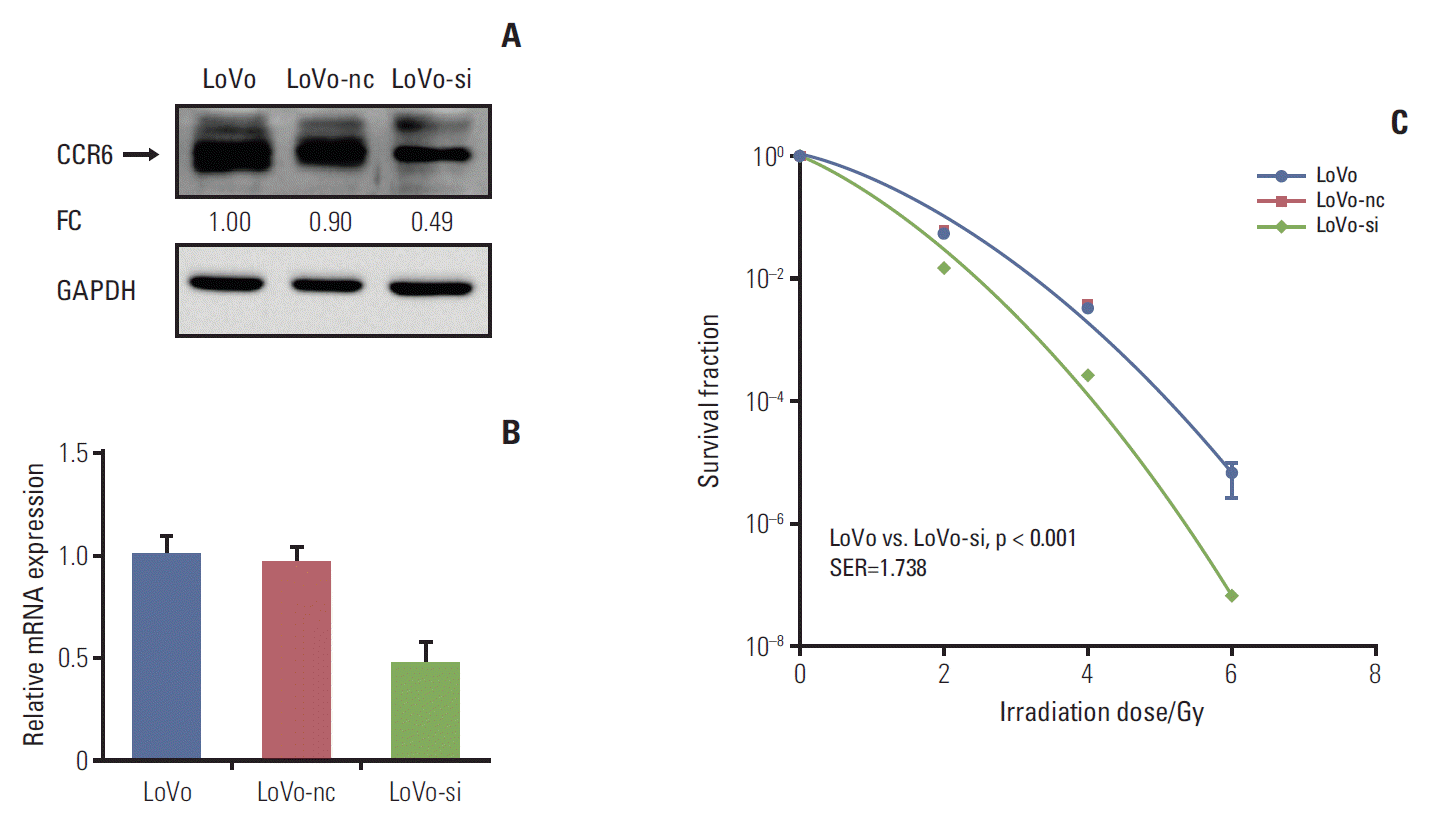
Fig. 4.
Inhibition of C-C motif chemokine receptor 6 (CCR6) expression improved radioresistance of colorectal cancer (CRC) cells. (A) Western blot analysis on CCR6 expression in the LoVo cells, the LoVo cells transfected with negative control (LoVo-nc), and the siRNA-transfected LoVo cells (LoVo-si). The reference cell line to calculate the fold change (FC) was the LoVo cell line. (B) Real-time polymerase chain reaction analysis on CCR6 expression in the CRC cell lines described in panel A. (C) Postirradiation survival curves of the CRC cell lines described in panel A. The LoVo-si cells was more sensitive to ionizing radiation than the LoVo cells. The sensitization enhancement ratio (SER) was 1.738 (p < 0.001). GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
![]()
4. Postirradiation DDR in CRC cells was retarded by CCR6 knockdown
The γH2AX is known to recognize the sites of DNA damage and initiate the DDR procedure. Therefore, the γH2AX has now been used as a practical molecular marker reflecting the existence of DNA damage [5,21]. Quantification of γH2AX-positive cells (in number per 100 cells) was conducted to decide the DDR abilities of CRC cell lines with different levels of CCR6 expression. Through IF analysis, the γH2AX-positive cells appeared to be similar among the LoVo, the LoVo-nc and the LoVo-si cell lines, at baseline and 30 minutes after 2 Gy irradiation. It indicated that irradiation caused similar DNA damage among the three cell lines. However, at 24 hours after 2-Gy irradiation, residual γH2AX-positive cells were significantly less in the LoVo (35.6±6.3 vs. 68.0±4.9, p=0.022) and the LoVo-nc (32.0±3.5 vs. 68.0±4.9, p=0.009) cell lines than in the LoVo-si cell line (Fig. 5). In other words, the DDR was retarded and more DNA damage remained in the LoVo-si cell line, in which CCR6 was knockdown. It implied that CCR6 might regulate radioresistance by affecting efficiency of IR-induced DDR.
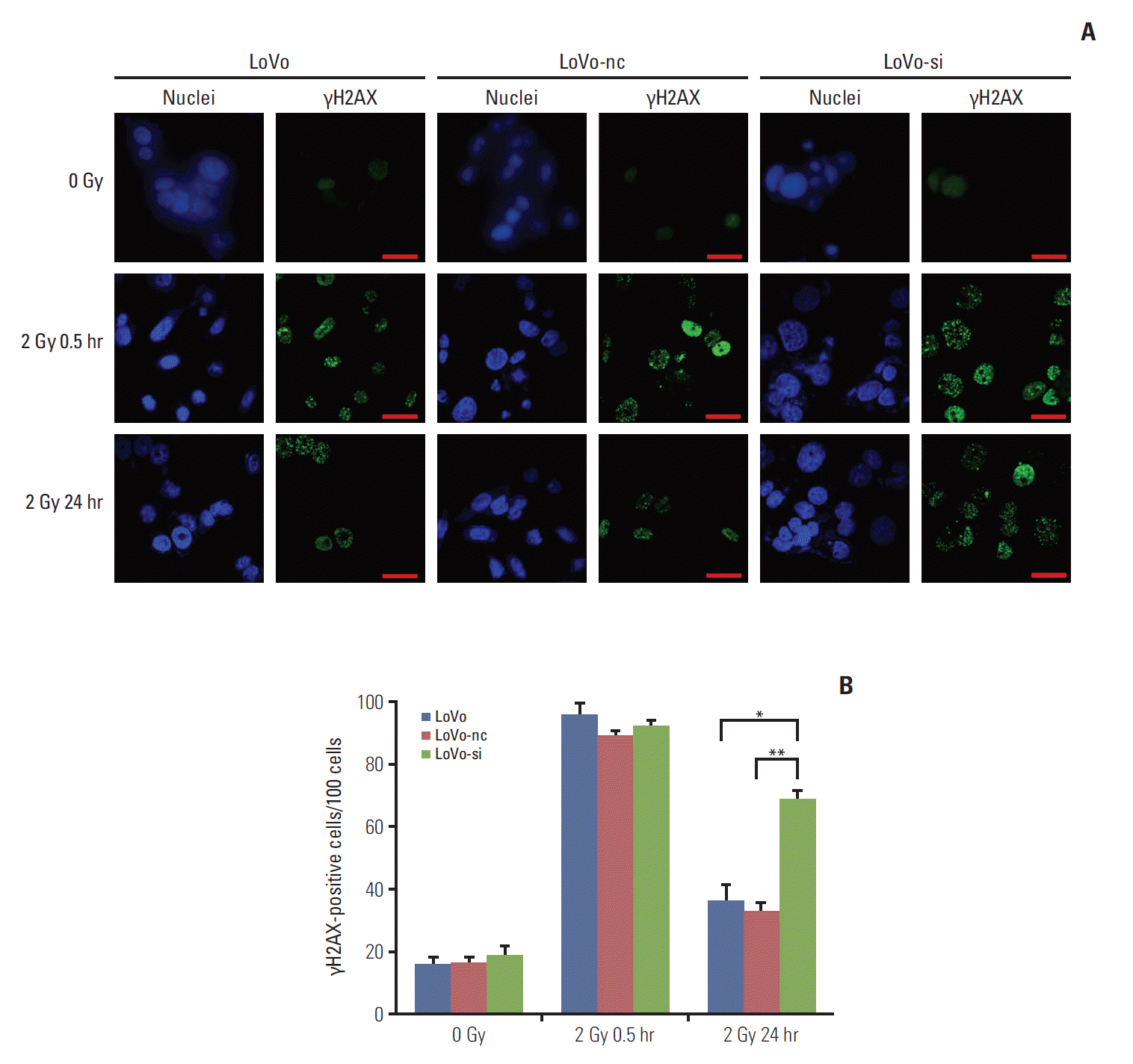
Fig. 5.
C-C motif chemokine receptor 6 knockdown resulted in retardation of postirradiation DNA damage repair in colorectal cancer (CRC) cells. (A) Immunofluorescent staining of nuclei and γH2AX in LoVo, LoVo cells transfected with negative control (LoVo-nc), and siRNA-transfected LoVo cells (LoVo-si) cell lines (×400). Each cell line was stained at 0 Gy, and at 30 minutes and 24 hours after 2-Gy irradiation. Scale bars=30 μm. (B) Quantification of γH2AX-positive cells (in number per 100 cells). Similar numbers of γH2AX-positive cells were seen among the three CRC cell lines described in panel A, at either 0 Gy or 30 minutes after 2-Gy irradiation. However, more γH2AX-positive cells were seen in the LoVo-si cell line than in the LoVo-nc and the LoVo cells lines at 24 hours after 2-Gy irradiation (*p < 0.05, **p < 0.01).
![]()
Discussion
The biological functions of CCR6 in metastasis and its predicting value in long-term outcome have been demonstrated in a series of studies. Chin et al. [22] showed in an in vitro study that activation of CCR6 could promote migration of CRC cells. And Kapur et al. [9] and Ghadjar et al. [8] further attained clinical evidences that high level of CCR6 expression would facilitate the distant metastasis of CRC, and was able to predict a poorer survival independently. Since therapeutic effect of RT is also so important for patients with locally advanced RC, impact of CCR6 on radiosensitivity of RC was explored in this study. It was found in RC patients that there was an inverse correlation between CCR6 expression and RT effect. Both the RNA-seq and the IHC analyses indicated that the lowlier CCR6 was expressed, the worse TRG would be got. Similar results were seen in CRC cell lines. Clonogenic survival assay after irradiation revealed that CCR6 expression was upregulated in cell lines (sw480 and LoVo) relatively resistant to IR. And on the contrary, down-regulation of CCR6 expression was seen in sw620, a radiosensitive cell line. Moreover, when CCR6 expression was interfered through siRNA, the LoVo cells would become sensitive to IR as well. These results all suggested that CCR6 might be a predicting biomarker of RT effect and a potential therapeutic target for reversing radioresistance. Prior to our study, there was no study focusing on the correlation between CCR6 expression and RC radiosensitivity. So, we are convinced the results might be informative for development of RT-sensitizing treatments and selection of individualized therapeutic strategies. Considering the proven influences of CCR6 on distant metastasis of CRC, the results of this study conferred more values on this biomarker, in improving prognosis of the RC patients.
The IR causes cell death mainly through double strand break of DNA. After irradiation, a tumor cell will survive if DDR is completed successfully. Or the programmed death will start instead. Therefore, the intrinsic resistance caused by DDR is acknowledged as the most critical mechanism of cellular radioresistance [5,23]. The mammalian DDR procedure is a phosphorylation signaling cascade. The first step is the phosphorylation on serine 139 of H2AX by ATM. The γH2AX will be dephosphorylated soon after DDR restores chromatin integrity [21,24,25]. Hence, the quantity of γH2AX is positively correlated with the number of DNA damage. We evaluated DDR abilities of different CRC cell lines after irradiation through quantifying the γH2AX-positive cells in this study. At 30 minutes after irradiation, an obviously increase of γH2AX-positive cells was seen in all the three tested cell lines, compared with the baseline. It indicated that DDR started as expected. And no difference in positive cell numbers was seen among the three cell lines, either before irradiation or at 30 minutes after irradiation. That was to say, similar amount of DNA damage was caused among the three cell lines. The numbers of γH2AX-positive cells then decreased at 24 hours after irradiation, indicating the gradual completion of DDR. But comparison among the three cell lines revealed that there was a delay of γH2AX clearance in the LoVo-si cell, unlike the LoVo and the LoVo-nc cells. It could be easily inferred that DDR was disrupted when CCR6 expression was interfered. Namely, CCR6 might play its part in regulation of radiosensitivity through affecting DDR efficiency.
Up to now, the exact mechanisms through which CCR6 regulates DDR remain uncertain. This regulation might lie on the Akt and the ERK pathways which could be activated by the combination of CCR6 and its sole ligand, CCL20 [11]. The activated Akt has been reported to stabilize the ubiquitin-conjugating enzyme E2S, which will subsequently recruit the Ku70, a key molecule in DDR process [12]. The Akt could also upregulate the expression of Rad51. The latter is a molecule participating in the DDR directly [13]. For ERK, it has been showed in a study of Marampon et al. [14] to upregulate the expression of the regulatory and catalytic subunits of the DDR complex, DNA-PK, after its activation. In addition, the Akt and the ERK have been reported to inhibit the programmed cell death through p53 and mammalian target of rapamycin pathways, which are activated when DDR fails to complete [26-28]. Further studies are in need to figure out whether CCR6 controls the DDR and radiosensitivity of CRC cells through these pathways.
More and more biological functions of the tumor cells, including resistance to IR or chemotherapy agents, has been discovered to be determined by the TME [6,29]. Chemokines receptors are important components linking the TME and the tumor cells. Through the pathway enrichment analysis on our RNA-seq data, the chemokine-mediated signaling pathway was showed as the fourth enriched pathways (p < 0.001) involved in RC radioresistance. Actually, before our study, another chemokine receptor, CXCR4, has been proven to regulate radiosensitivity of glioblastoma and uterine cervical cancer [30]. The results of our study added knowledge to this field and might help radiation oncologists to design further researches to explore and interfere the relationship between TME and the radioresistence of malignant tumors. Furthermore, the modalities blocking the interaction between chemokines and their receptors have gradually been developed [29]. The chemokine receptors are expected to be practical targets for radiosensitization treatment.
In conclusion, this study demonstrated that CCR6 overexpression contributed to a poorer RT effect in RC patients and radioresistance in CRC cells. And the results also implied that the CCR6 might enhance radioresistance through more effective DDR. Thus, inhibition of CCR6 expression might be a therapeutic strategy to sensitize RC to RT.
 XML Download
XML Download