

 PDF
PDF Citation
Citation Print
Print


Introduction
Glioblastoma multiforme (GBM) is the most prevalent and deadly brain tumor, with an average survival of less than 1.5 year. The current treatment regimen includes surgery, radiotherapy, and chemotherapy. Temozolomide (TMZ) is one of the major components of chemotherapy regimens. However, despite multimodal therapies most patients suffer recurrence and die within 40 weeks [1,2]. There is no consensus on the treatment for recurrent and TMZ-refractory GBM. Various combination therapies with TMZ have been investigated for newly diagnosed and recurrent GBM [3-5].
Recent work has shown that metformin, an anti-diabetes agent, exhibited anti-cancer effects in a variety of tumors including breast cancer, pancreatic cancer, colon cancer, and ovarian cancer [6-9]. In addition, metformin might have synergistic effects with TMZ treatment and can enhance chemotherapy efficacy in GBM [10-12], opening a new avenue to overcoming TMZ resistance in glioma treatment. As an inexpensive, well-tolerated, first-line oral anti-diabetes drug, metformin suppresses hepatic glucose production and reduces insulin resistance in peripheral tissues. In addition, metformin also functions through the fatty acid metabolism pathway by de-repressing fatty acid oxidation [13].
Several mechanisms have been reported to be involved in the anti-cancer effects of metformin. Previous studies have demonstrated that metformin activates the AMP-activated protein kinase (AMPK)‒mammalian target of rapamycin (mTOR) signaling pathway, which is also involved in the regulation of cancer cell survival, proliferation, and apoptosis, as well as the epithelial-to-mesenchymal transition phenotype [7-9].
In this study, we showed the potential efficacy of combined treatment with metformin and TMZ in Vitro. This efficacy was confirmed in vivo in a xenograft mouse model of glioblastoma treated with clinically relevant dosages of metformin and TMZ. Additionally, the efficacy of high-dose metformin combined with TMZ was investigated to evaluate the possible translational value for clinical applications.
Materials and Methods
1. Cell culture
Human glioblastoma cell lines U87 and A172 originally obtained from American Type Culture Collection (ATCC) and U-251 MG originally obtained from Sigma-Aldrich (St. Louis, MO) were cultured in Eagle's minimum essential medium supplemented with 10% fetal bovine serum, 1% MEM NEAA (Life Technologies, Carlsbad, CA), and 1% GlutaMAX (Life Technologies) at 37℃ and 5% CO2 in a humidified incubator. TMZ (Sigma-Aldrich) was dissolved in dimethyl sulfoxide to prepare a stock concentration of 200 mM, which was further diluted in cell culture medium to working concentrations.
2. Cell viability assay
Glioblastoma U87, U251, and A172 cells (1×104 cells/well) were plated in 96-well flat bottom tissue culture plates and incubated at 37℃ in a 5% CO2/95% air atmosphere. The cells were treated for 24, 48, and 72 hours with 50, 250, and 500 μM TMZ or for 24, 48, and 72 hours with 5, 10, and 20 mM metformin (Sigma-Aldrich). Next, combined administration of TMZ and metformin was performed in the same manner. After treatment for 24, 48, or 72 hours, 10 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) stock solution (Ez CyTox, Daeil Lab Service Co., Ltd., Seoul, Korea) was added to each well, and the plates were incubated for 4 hours. Plates were agitated on a plate shaker for 3 seconds, and the absorbance at 540 nm was determined using a scanning multi-well spectrophotometer (VERSA max, Molecular Device, Sunnyvale, CA) and cell viability (%) was determined by normalizing each group to the control.
3. Apoptosis assay
U87 cells were plated in 12-well plates and treated with TMZ (50, 250, and 500 μM), metformin (5, 10, and 20 mM), or a combination of TMZ and metformin for 48 hours. After treatment, the cells were washed and allowed to grow in TMZ-free medium for 48 hours. The apoptosis ratio was analyzed using an Annexin V FITC Apoptosis Detection Kit (BD Biosciences, San Diego, CA) according to the manufacturer's instructions. Annexin V/FITC and propidium iodide double staining was used to evaluate the percentages of annexin V–/propidium iodide (PI)+ (necrosis), annexin V+/PI– (early apoptosis), and V+/PI+ (later apoptosis) cells. Tests were repeated in triplicate.
4. Intracranial inoculation of cancer cells and experimental design
Athymic nude mice were anesthetized with an intraperitoneal injection of 12 mg/kg xylazine (Rompun, Cutter Laboratories, Shawnee, KS) and 30 mg/kg ketamine (Ketalar, Parke-Davis & Co., Morris Plains, NJ). The mice were then stereotactically inoculated with 5×105 U87 cells into the right frontal lobe (2 mm lateral and 1 mm anterior to bregma, at a depth of 2.5 mm from the skull) using a sterile Hamilton syringe fitted with a 26-gauge needle (Hamilton Co., Reno, NV) and a microinfusion pump (Harvard Apparatus, Holliston, MA).
Each experimental group contained five mice. Mice in the first group were treated with metformin (2 mg/25 g/day) via intraperitoneal injection for 4 weeks after intracranial inoculation with U87 cells. Mice in the second treatment group were treated with TMZ (15 mg/kg/day) via intraperitoneal injection for 4 weeks after intracranial inoculation. Mice in the combination treatment groups were treated with metformin (2 mg/25 g/day or 10 mg/25 g/day) and TMZ (15 mg/kg/day) via intraperitoneal injection for 4 weeks.
5. Western blot analysis
Total protein was extracted using a PhosSTOP EASYpack (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and detected with antibodies against p53, AMPK, mTOR, fatty acid synthase (FASN) (Cell Signaling Technology, Danvers, MA), and β-actin (Sigma). Immunoreactivity was detected using the ECL chemiluminescence system and quantified using an imaging densitometer. The density of each band was quantified using Quantity One software (Bio-Rad, Hercules, CA).
6. Immunofluorescence analysis
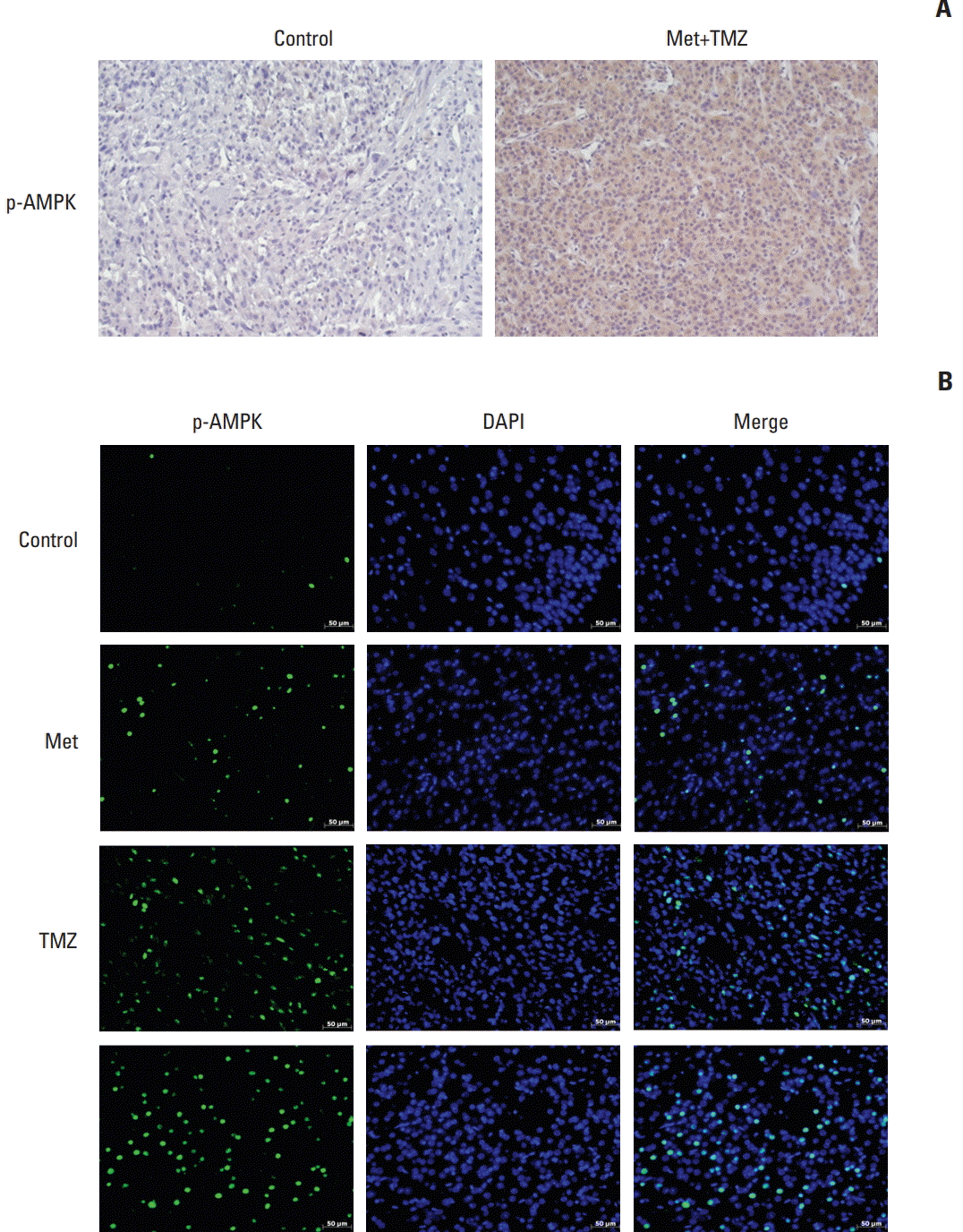
We performed immunofluorescence analysis for phospho-Thr172 AMPK (1:25, Cell Signaling Technology) in brain tissue sections using a tyramide signal amplification fluorescence system (Perkin Elmer, Waltham, MA). The samples were counterstained with 4,6-diamidinO2-phenylindole (DAPI). Fluorescent images were examined under a laser scanning confocal microscope system (LSM 700, Carl Zeiss, Oberkochen, Germany).
7. Statistical analysis
The results of cell survival assays were analyzed by a twotailed Student's t test. Overall survival was analyzed using the Kaplan-Meier method, and survival data were compared using a log-rank test. A p-value of < 0.05 was considered statistically significant. Statistical analysis was performed with the SPSS ver. 23.0 (IBM Corp., Armonk, NY).
Results
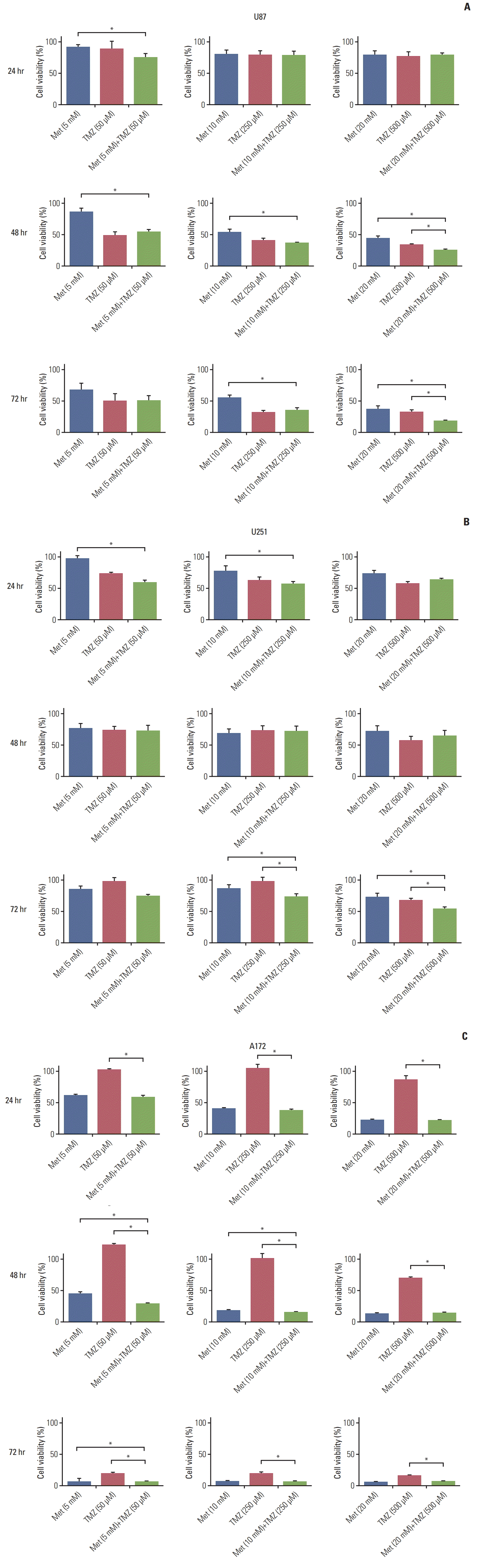
1. MTT assay
We performed the MTT assay to determine the combination effect of TMZ and metformin in U87, U251, and A172 cell lines. The combination of TMZ (50 μm) and metformin (5 mM) did not show higher cytotoxicity against U87 cells than TMZ (50 μm) only or metformin (5 mM) only after 72-hour treatment. The combination of TMZ (250 μm) and metformin (10 mM) showed higher cytotoxicity against U87 cells than metformin (10 mM), but not TMZ (250 μm) after 48-hour and 72-hour treatment. The combination of TMZ (500 μm) and metformin (20 mM) showed higher cytotoxicity against U87 cells than TMZ (500 μm) only or metformin (20 mM) only after 48-hour and 72-hour treatment. A significant additive effect was not seen after 24-hour or 48-hour treatment in U251 cells. The combination of TMZ (250 and 500 μm) and metformin (10 and 20 mM) showed higher cytotoxicity against U251 cells than TMZ only or metformin (20 mM) only after 72-hour treatment. The combination of TMZ (50 μm) and metformin (5 mM) showed higher cytotoxicity against A172 cells than TMZ (50 μm) only or metformin (5 mM) only after 48-hour and 72-hour treatment. The combination of TMZ (500 μm) and metformin (20 mM) showed higher cytotoxicity against A172 cells than TMZ (500 μm), but not metformin (20 mM) after 48-hour and 72-hour treatment. Overall, the combination of TMZ (250 μm) and metformin (10 mM) showed the highest cytotoxicity after 48-hour treatment (Fig. 1).
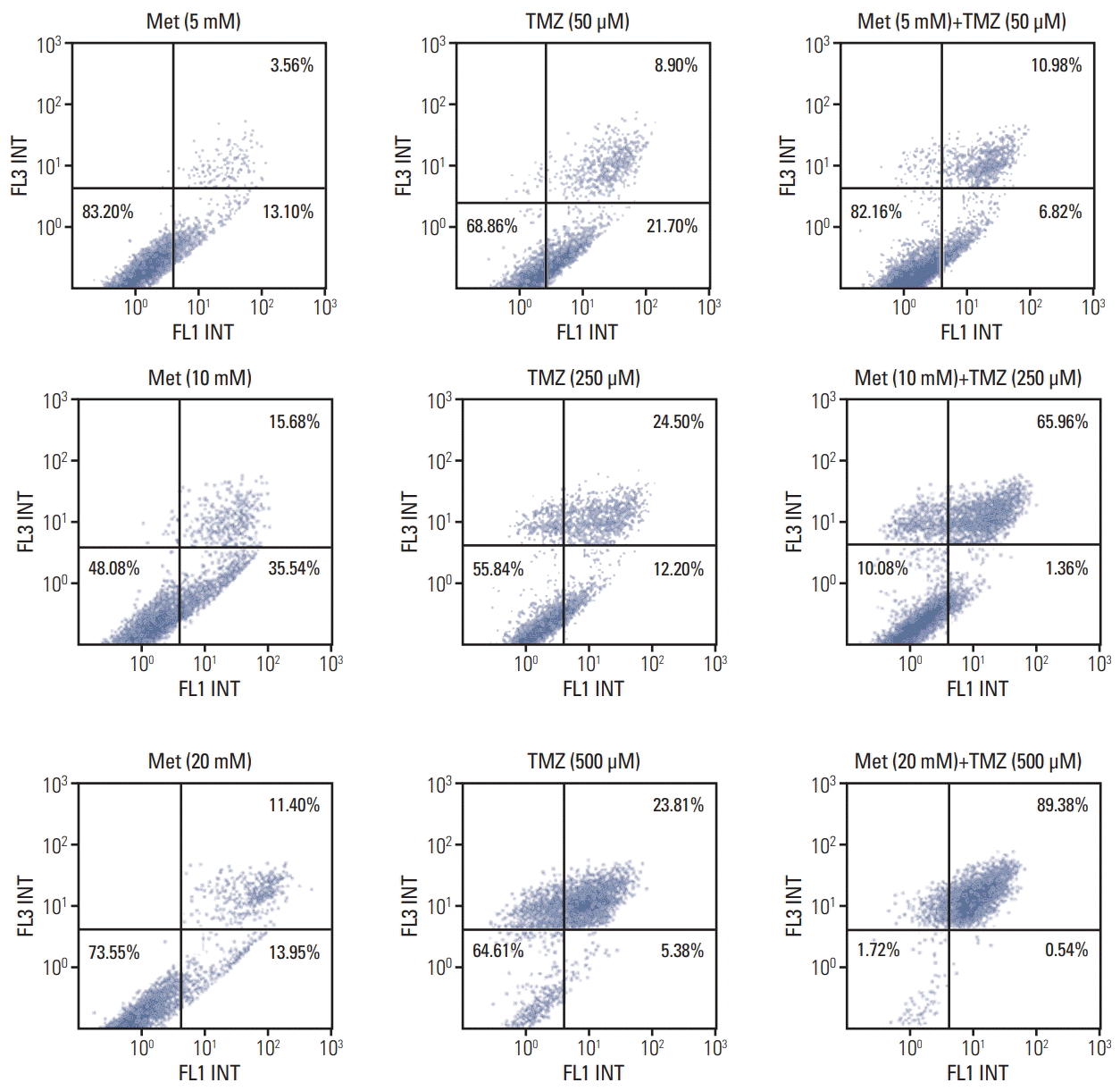
2. Fluorescence-activated cell sorting analysis
We next determined whether the TMZ- or metformininduced reduction in cell viability was accompanied by cell apoptosis. Annexin V/PI staining with flow cytometry was used to detect apoptosis in U87 cells treated with TMZ (5, 10, and 20 mM), metformin (50, 250, and 500 μM), or a combination of TMZ and metformin for 48 hours. As shown in Fig. 2, combined treatment with TMZ and metformin induced a higher level of cell apoptosis with increasing doses of TMZ and metformin, respectively. The high-dose combination of metformin (20 mM) and TMZ (500 μM) showed the highest apoptotic activity.
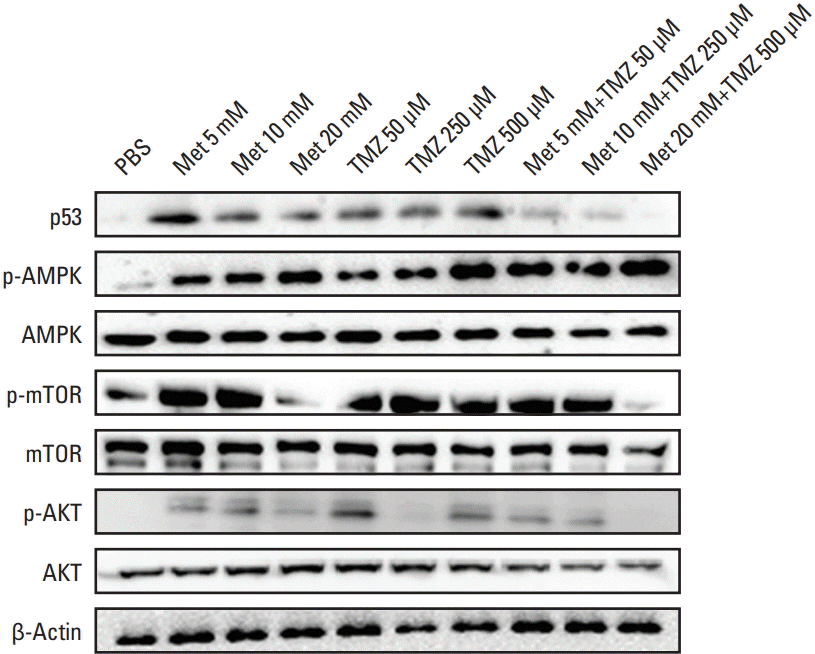
3. Western blot and immunofluorescence analysis
Metformin, as an AMPK-activating agent, is widely used to suppress tumor cell proliferation. To examine whether the growth inhibitory effect of treatment with TMZ and metformin in GBM was mediated by activation of the AMPK signaling pathway, we performed western blot analysis of AMPK, mTOR, AKT, and p53 expression in U87 cells treated with metformin, TMZ, or the combination of metformin and TMZ.
As shown in Fig. 3, both TMZ and metformin clearly induced AMPK phosphorylation in a dose-dependent manner. The combination of TMZ and metformin enhanced AMPK phosphorylation. High-dose metformin (20 mM) inhibited mTOR and AKT phosphorylation. Although TMZ treatment did not affect mTOR phosphorylation, the combination treatment of TMZ and metformin significantly inhibited mTOR phosphorylation and p53 expression. It could be caused by the effect of high-dose metformin. Activation of phosphorylated AMPK and its downstream molecules was greater under combination treatment compared with singledrug treatment.
4. Glioblastoma mouse model
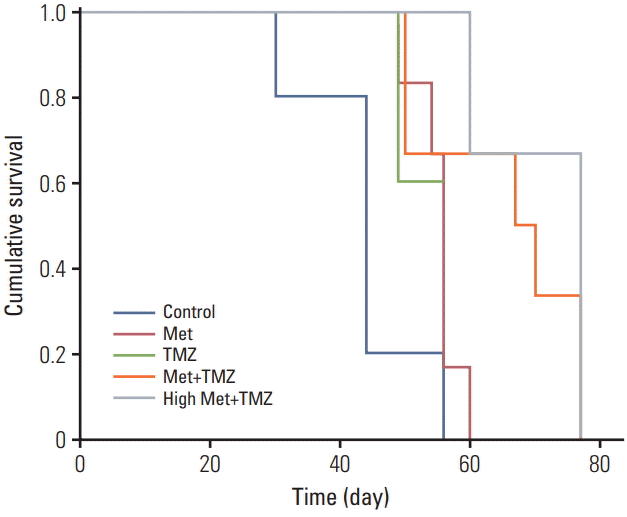
We also examined whether the combination of TMZ and metformin displays synergistic anti-glioma effects in vivo. Mice in the combination treatment group were treated with metformin (2 mg/25 g/day) and TMZ (15 mg/kg/day) via i.p. injection for 4 weeks. These dosages are clinically relevant [14,15]. Additionally, we compared the efficacy of combined treatment with a higher dosage of metformin (10 mg/25 g/day) and TMZ (15 mg/kg/day) (S1 Fig.).
The median survival of each group was 43.6, 55.2, 53.2, 65.2, and 71.3 days for control, metformin treatment (2 mg/25 g/day), TMZ treatment (15 mg/kg/day), combination treatment with low-dose metformin (2 mg/25 g/day) and TMZ (15 mg/kg/day), and combination treatment with high-dose metformin (10 mg/25 g/day) and TMZ (15 mg/kg/day), respectively (Fig. 4). Combination treatment with high-dose metformin and TMZ is superior to monotherapy (p < 0.05) but is similar to low-dose metformin and TMZ (p=0.43) in terms of survival benefit.
AMPK immunohistochemistry was strongly positive in mice treated with the combination of metformin and TMZ compared with the control group. Similarly, immunofluorescence showed higher expression of AMPK in the tumors treated with the combination of metformin and TMZ (Fig. 5).
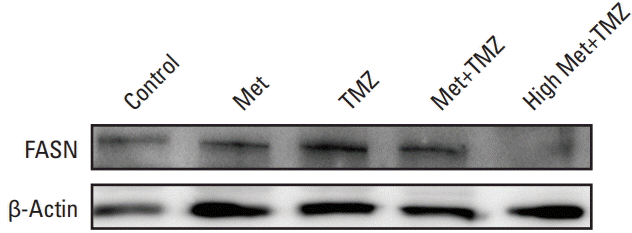
FASN expression was investigated to explain the mechanism by which the combination treatment with high-dose metformin was superior to combination treatment with the clinically relevant dosage of metformin. The expression of FASN was significantly decreased in tumor specimens treated with metformin (10 mg/25 g/day) and TMZ (15 mg/kg/day) (Fig. 6).
Discussion
In this study, we showed that combination treatment with metformin and TMZ was superior to monotherapy using metformin or TMZ in terms of cell viability in Vitro and survival in vivo. The action mechanism of metformin on cancer cells involves cytotoxicity mediated through AMPK-p53-mTOR pathway. Our results revealed that combined treatment of TMZ and metformin enhanced the activation of the AMPK-p53-mTOR pathway compared to monotherapy with either TMZ or metformin. Superiority of the combination strategy of metformin and TMZ has been reported recently in two studies [11,12]. However, one study was restricted to in Vitro experiments only [11]. The other study showed efficacy of the combination treatment in vivo using a model in which 1×106 U87 cells were injected subcutaneously into the right flank of immunodeficient mice [12]. Combined treatment of metformin (400 mg/kg/day) and TMZ (25 mg/kg/day) significantly reduced tumor growth rates and prolonged survival in in vivo xenograft models. They found that combined treatment of metformin and TMZ synergistically inhibited proliferation and induced apoptosis of both glioma cells and glioma stem cells (GSCs) through downregulating the AKT-mTOR signaling pathway. In the present study, we established an orthotopic GBM mouse model and administered clinically relevant dosages of metformin and TMZ. We believe that our protocol is more similar to clinical settings than previous studies. in vivo results following treatment with clinically relevant dosages of both metformin and TMZ showed a survival benefit compared to monotherapy. We also investigated the efficacy of a higher dosage of metformin (five times higher than the initial dose). A trend of survival gain was noted with the higher dose, although the survival gain was not statistically significant. More importantly, mice tolerated the high dose of metformin. To the best of our knowledge, only three clinical trials have been undertaken using metformin to treat malignant glioma in adults (NCT-02149459, NCT02780024, and NCT01430351). The dosage of metformin administered is up to 2,000 mg a day, comparable to that administered for diabetic patients. In a phase 2 trial to access the efficacy after the addition of metformin to standard therapy in patients with advanced pancreatic cancer, patients with high plasma concentrations (> 1 mg/L) of metformin seemed to have improved survival [16]. Patients with diabetes given metformin at doses of more than 2,000 mg per day could develop unacceptable toxic effects. Therefore, simple escalation of metformin dosage is unlikely to be clinically feasible for treating GBM patients. Further studies are needed to test more potent biguanides because a higher dose of metformin might activate the AMPK-p53-mTOR pathway and provide survival benefit.
FASN is a multifunctional enzyme that plays a central role in fatty acid synthesis and lipid biosynthesis [17]. In various cancers, aggressive features such as migration, invasion, metastasis, and chemo-resistance have been shown to be dependent on FASN [18-20]. Overexpression of FASN is also associated with glioma grade. Treating glioblastoma cells with FASN inhibitors has resulted in significant reduction in tumor cell viability [21]. It has been reported that inhibition of FASN can block hypoxia-inducible factor-1α/vascular endothelial growth factor A (VEGF-A) signaling and upregulate anti-angiogenic isoform-VEGF165b [22]. FASN inhibition has markedly decreased the proliferation and migration of GSCs, although the mechanism by which FASN-mediated cellular fatty acid homeostasis regulates the biological features of GSCs is currently unknown [23]. One author of the present study has recently reported that upregulated FASN expression in TMZ-resistant lines is decreased after metformin treatment [24]. We compared expression levels of FASN between treatments using clinically relevant dosage and high dosage of metformin. Interestingly, FASN expression was decreased to a greater extent after treatment with high-dose metformin. Lipid metabolism might be considered as a new therapeutic target for GBM. Many cytotoxic chemotherapy drugs and cytostatic targeted agents have failed to increase the survival of GBM patients. Metabolic targeting using metformin could be an alternative strategy for newly diagnosed GBM and recurrent GBM. Our study provides further evidence that metformin is effective for treating GBM. However, its optimal treatment dosage and duration remain to be determined through further studies. In addition, FASN inhibition by metformin should be validated with patient-derived models. More potent drugs targeting the AMPK-p53-mTOR pathway are also needed for treating GBM.
The combination of metformin and TMZ has superiority over monotherapy using metformin or TMZ in terms of GBM cell viability in Vitro and survival in vivo. The combination of high-dose metformin and TMZ inhibited FASN expression. The present data confirm that metformin might be a good candidate for a combined regimen with TMZ in clinical settings.
 XML Download
XML Download