

 PDF
PDF Citation
Citation Print
Print


Introduction
Glioma are categorized according to their grade based on histological and pathological features based on World Health Organization (WHO) classification. Glioblastoma (GBM, WHO grade IV glioma) is the most aggressive malignant primary brain tumor [1,2]. Current standard regimen consists of surgical resection followed by radiation and chemotherapy [3,4]. Despite such aggressive treatment option, the average survival of GBM patients spans only 1-2 years [4], requiring a new therapeutic approach.
Dynamic interaction between ligand and its partner receptor regulates essential cellular functions including cell survival, proliferation, and migration [5]. In cancerous tissues, including GBM, a multitude of ligand-receptor signaling axis are frequently dysregulated, resulting in tumor malignancy and dismal prognosis [6]. Recent large-scale genomic studies have revealed a catalog of core oncogenic pathways that are frequently activated in tumor progression including receptor tyrosine kinases that encompass epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor, and c-MET [7,8]. As tumor cells often depend on oncogenic signaling pathways for survivability, inhibition of these pathways has been recognized as a potential therapeutic approach. Antibody-mediated therapy has been a promising approach, as shown by its clinical success involving anti-EGFR antibodies [9]. Currently, 13 Food and Drug Administration approved antibodies have been translated into a clinical course and newly developed antibodies are currently being evaluated in various stages of clinical trials [10].
Semaphorins are able to regulate cell to cell interaction and differentiation [11]. There are 20 types of mammalian semaphorins which are categorized into five classes (classes 3-7) according to their molecular structures [12]. Among five classes, only class 3 Semaphorins are secreted proteins, whereas the rest are produced as membrane-anchored or transmembrane proteins [13]. Semaphorin3A (SEMA3A) is widely known as an axon guidance factor; however, recent studies have demonstrated its oncogenic roles in various cancer types [14-16]. In pancreatic cancer, SEMA3A promotes dissemination and invasion through activation of multiple pathways involving Rac1, GSK3, and p42/p44 mitogen-activated protein kinase (MAPK) [17]. In addition, autocrine SEMA3A in GBM promotes cell dispersal by modulating substrate adhesion and elevates vascular permeability, resulting in tumor-promoted vascular leakage in vitro and in vivo [18,19]. SEMA3A in hepatocellular carcinoma functions as a chemoattractant involved in tumor-associated macrophages (TAM) infiltration and promotes tumor proliferation and migration [20]. These studies suggest SEMA3A as a potential therapeutic target in suppressing cancer proliferation and invasion. However, development of SEMA3A targeting antibody has not been initiated. In present study, we generated fully human antibody that targets SEMA3A. Phage display technology has been widely employed for high-throughput generation of antibodies [21,22]. Through a single chain fragment variant (scFv) phage display screen, we isolated anti-SEMA3A scFvs. Using the Expi293F Expression System (Thermo Fisher Scientific, Waltham, MA), we established three different fully human anti-SEMA3A IgG antibodies and determined their therapeutic efficacy in GBM.
Materials and Methods
1. Statistical analysis
Expression profiling of Repository of Molecular Brain Neoplasia Data (REMBRANDT) glioma dataset were downloaded as CEL files. Expression levels of each sample were summarized per each gene using Affymetrix U133 Plus 2.0 array annotation file (Affymetrix, Santa Clara, CA). SEMA3A mRNA expression levels by WHO glioma grade were statistically compared via student t test. For The Cancer Genome Atlas (TCGA) GBM data set, Kaplan-Meier survival analysis was available from cBioPortal (http://www.cbioportal.org/).
2. Screening of anti-SEMA3A scFv
We performed selection of SEMA3A-binding phage scFv via classical panning method using synthetic human scFv phage library. The antigens that were used for panning were recombinant human SEMA3A (rhSEMA3A)/Fc chimeric protein (R&D Systems, Minneapolis, MN) and Erbitux (Eli Lilly, Indianapolis, IN). Immunotubes (Nunc, Rochester, NY) were coated with 5 μg/mL of rhSEMA3A/Fc chimeric protein (R&D Systems) in phosphate buffered saline (PBS) for 1 hour at 37℃. After blocking the tube with 3% skim milk/PBS, Erbitux was added and incubated for 1 hour in room temperature during the binding step to eliminate scFvs that specifically bind to the Fc portions of rhSEMA3A/Fc. Bound phage-scFvs were eluted from the tube by adding 1 mL of 100 mM triethylamine and neutralized by adding 0.5 mL of 1.0 M Tris-HCl (pH 7.4). For the rescue step, eluted phagescFvs were infected into 10 mL of Escherichia coli, ER2537 cells in Super Broth medium.
3. Production and purification of anti-SEMA3A IgG
The Expi293F expression system (Thermo Fisher Scientific) was used for anti-SEMA3A IgG production. Using HiTrap protein G HP (5 mL) of ÄKTA Protein Purification System (GE Healthcare, Barrington, IL) purified anti-SEMA3A antibodies were concentrated by Amicon Ultra Centrifugal Filter (Merck Millipore, Burlington, MA). The produced anti-SEMA3A IgGs were evaluated by size exclusion high-performance liquid chromatography analysis and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for aggregation and degradation. in vivo experiment was further tested by LAL endotoxin quantitation kit (QCL-1000, Lonza, Basel, Switzerland).
4. ELISA
The flat bottom 96-well plates (Costar, Corning, New York, NY) were coated with 1 μg/mL of human, mouse recombinant protein (R&D Systems), Erbitux (IMC-C225), and bovine serum albumin (BSA; NEB, Ipswich, MA) overnight at 4℃. Plates were washed with PBST (0.1%) and blocked with 3% skim milk (BD Difco, Franklin Lakes, NJ) solution. The anti-SEMA3A scFvs and IgGs were incubated for 1 hour in room temperature. After washing with PBST (0.1%), anti-HA antibody conjugated to horseradish peroxidase (HRP; scFv) and the anti-human Fab antibody conjugated to HRP (IgG) were added for 1 hour in room temperature. Following washing step, each well was detected with tetramethylbenzidine (TMB) solution as HRP substrate for 5-25 minutes. The stop solution for TMB reaction was added and enzymelinked immunosorbent assay (ELISA) plate was analyzed at 450 nm.
5. Patient-derived GBM specimens
GBM specimens were obtained from patients undergoing surgery based on consent in accordance with the appropriate Institutional Review Boards. Patient-derived GBM cells were cultured in the Neurobasal-A medium (NBA) [23].
6. Proliferation assay
Using a 96-well plate (Corning), GBM patient cells (5×103 cells/well) were seeded into 100 μL of NBA medium and SEMA3A IgG or control human IgG (Thermo Fisher Scientific) were given for 3 days. After 6 and 9 days, 10 μL of EZ-Cytox (Daeil Lab, Seoul, Korea) was added to the plates and incubated for 2 hours. The optical density was measured at 450 nm using a microplate reader (Bio-Rad, Hercules, CA). The cells were plated in quintuplicate.
7. Migration assay
Migration assays were performed using the Transwell system, in which the upper chambers of the transwells were coated with poly-L-ornithine for 1 hour and dried. 1×105 GBM cells in 100 μL of media without any growth factors were seeded into the upper chambers and the lower chamber was filled with 600 μL of medium including growth factors. The anti-SEMA3A antibodies (scFv, 50 μg/mL; IgG, 10 μg/mL) were added into the upper chambers and lower chambers and transwells were incubated for 24 hours at 37℃. Next day, migrated cells on the lower surface of the upper chamber were fixed with methanol and stained with hematoxylin and eosin. The numbers of migrated cells were counted in eight different regions of the chambers that were selected randomly. The Oris cell migration assay (Platypus Technologies, Fitchburg, WI) was processed to evaluate anti-SEMA3A antibody effect in a dose-dependent manner and U87-MG (5×103 cells/well) cells were seeded in a 96-well plate. After 24 hours, migrated cells were quantified with Calcein AM fluorescent dye stained.
8. Subcutaneous xenograft tumor models
Patient-derived cells (PDCs) were injected into the subcutaneous region of 6-week-old female BALB/c nude mice. The tumor bearing mice were regrouped when mass volume reached at average of 80 mm3. Anti-SEMA3A antibody F11 and PBS were administered via intravenous injection. Mice with body weight loss greater than 20% were sacrificed.
9. Immunofluorescence analysis
Subcutaneous tumors were fixed in 5% paraformaldehyde and embedded in paraffin. Serial sections were stained with anti-SEMA3A (Abcam, Cambridge, MA) or anti-pERK (Cell Signaling Technology, Danvers, MA). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays were performed using the TUNEL apoptosis detection kit (Millipore, Billerica, MA). To evaluate TAM accumulation, paraffin sections were stained with anti-Iba1 (Abcam) as a macrophage marker.
10. Ethical statement
The study was approved by the Institutional Review Board of Samsung Medical Center (IRB No. 2010-04-004) and performed in accordance with the principles of the Declaration of Helsinki. Written informed consents were obtained.
All mouse experiments were performed according to the Association for Assessment and Accreditation of Laboratory Animal Care-accredited guidelines of our institute’s Animal Use and Care Committee.
Results
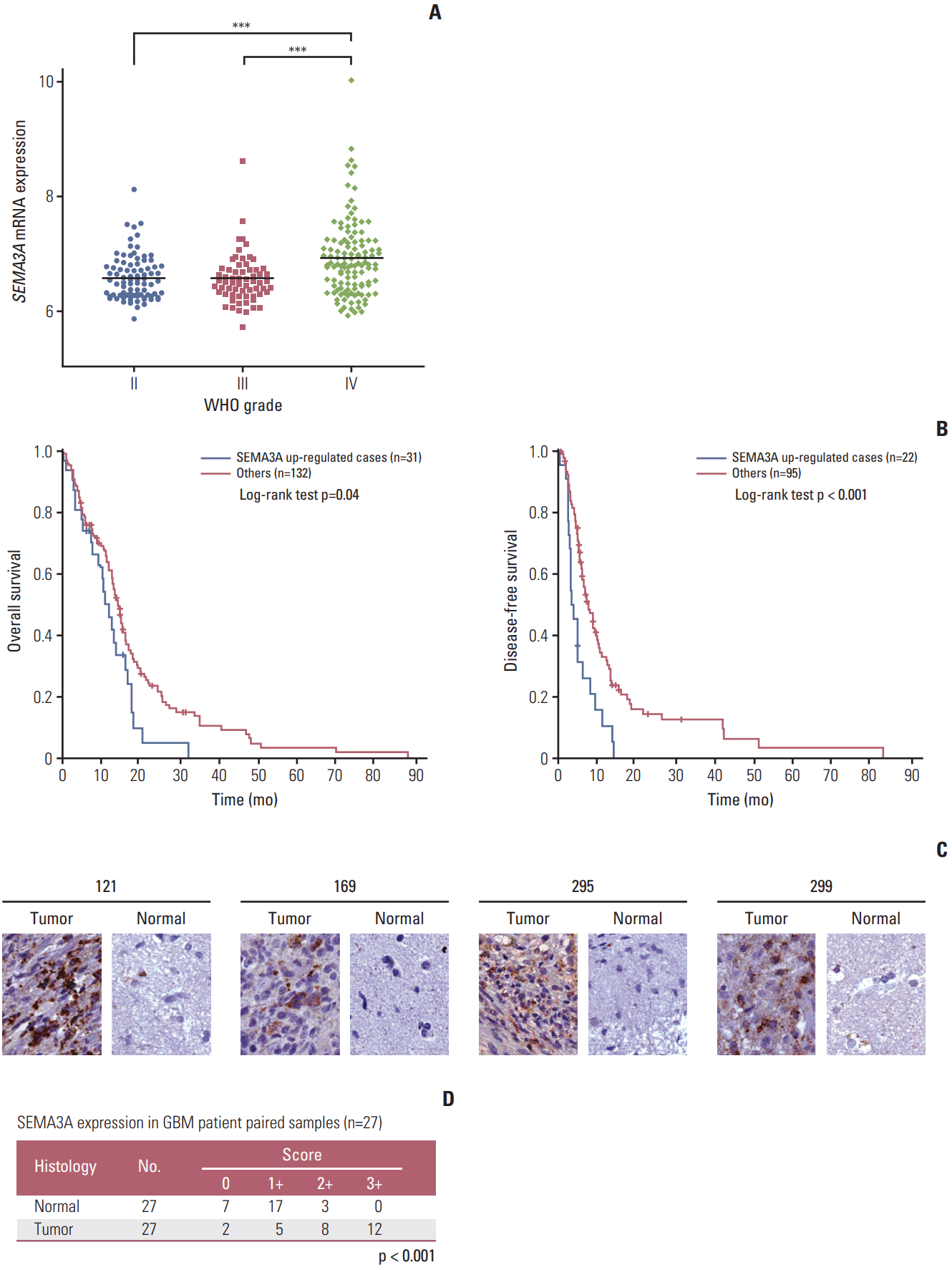
1. SEMA3A is highly expressed in GBM specimens compared to the normal brain tissues
We first analyzed the mRNA expression levels of SEMA3A in gliomas according to their pathological grade (II to IV) using Rembrandt glioma dataset. SEMA3A expression was significantly correlated with the glioma grade level. GBM, in particular, exhibited higher expression levels of SEMA3A compared to grade II and III gliomas (Fig. 1A). Overall survival and progression-free survival of patients in TCGA showed that the SEMA3A high expression group portrayed worse survival rate (p=0.04 and p < 0.001) (Fig. 1B). Our results suggest that upregulation of SEMA3A expression is directly correlated with disease progression in GBM. To determine the SEMA3A protein level in GBM specimens, we conducted tissue microarray analysis on 27 GBM and matched adjacent non-neoplastic brain tissue samples. Immunohistochemical analysis showed elevated protein levels of SEMA3A in all GBM tissue samples, while the majority of non-tumor tissues showed minimal levels of SEMA3A (Fig. 1C and D). Collectively, the malignant effect of SEMA3A expression in GBM specimens is evident and SEMA3A appears to be an ideal therapeutic target in GBM treatment.
2. Identification of SEMA3A-specific scFv using synthetic phage library
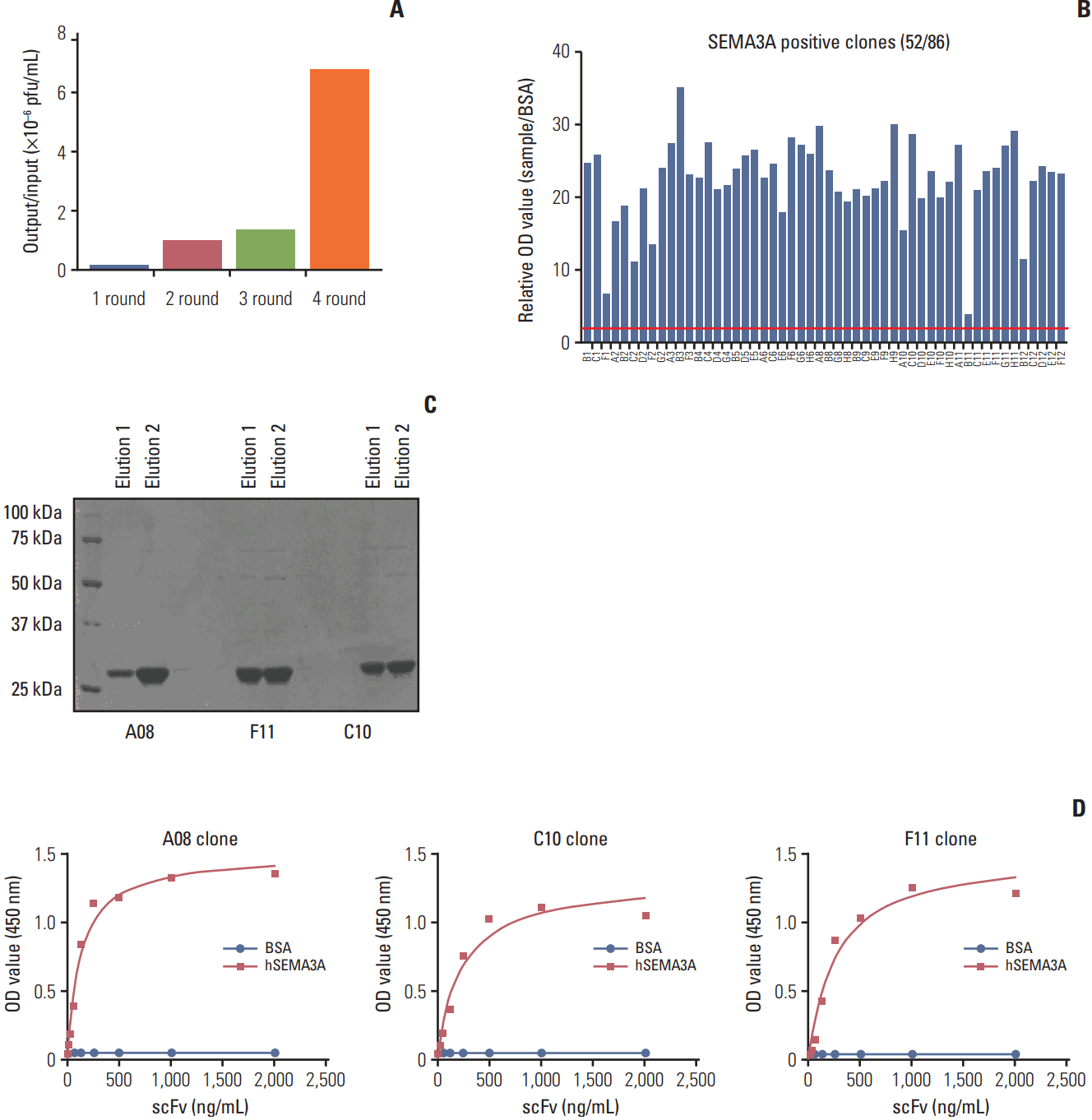
In present study, we generated a scFv with high affinity to SEMA3A using a synthetic human scFv phage library. The synthetic human scFv phage library with a total diversity of 7.6×109 cfu were composed of six diversified complementarity determining regions (CDRs) [24]. We performed a classical panning method using immunotube-immobilized rhSEMA3A/Fc chimeric protein. In order to prevent any scFvs from binding to the Fc portion, we combined our synthetic scFv library with Erbitux containing the Fc region and proceeded on to the selection process. Approximately 70-fold increase in output to input phage ratio was observed after the fourth selection round compared to the first, indicating the specificity of the accumulated phages to human SEMA3A (hSEMA3A) (Fig. 2A).
We incubated the final resulting outputs on Luria-Bertani/ampicillin plate in 37℃ overnight. Afterwards, we randomly picked 86 different single colonies and conducted phage ELISA. In accordance with the given criteria (at least two-fold increase to the negative control, i.e., BSA), 52 out of 86 colonies (60%) were identified as rhSEMA3A binders (Fig. 2B). Erbitux was also used as a negative control (data not shown). Through sequence analysis of the 49 positive clones, we obtained three final clones that were specific for SEMA3A. Our final candidates, F11, A08, and C10, possessed specific sequences that are identical to other 22, 4, and 3 other clones, respectively. To produce purified forms of the generated scFv, we inserted our three clones into E. coli Top10F, which recognizes the amber codon residing in the front of gene III and removes it in process. The resulting purified clones were verified through denaturation using Coomassie blue staining method (Fig. 2C). As the size of a typical scFv is at approximately 28 kDa, we expected our clones to appear between 25 and 37 kDa markers. As we used PBS with 200 mM imidazole as our elution, eluted anti-SEMA3A scFv were processed under SDS-PAGE analysis without the imidazole. To examine the binding activity of the anti-SEMA3A scFvs, we measured the binding affinity of each clone through indirect ELISA analysis with at serial antibody concentration levels. We used as 12B clone as negative control scFv having no binding to human Sema3A (S1 Fig.). Our results showed that the binding activity of each anti-SEMA3A clone to hSEMA3A protein was elevated in a dosedependent manner, while BSA, the control, displayed no significant changes (Fig. 2D). Overall, we have successfully produced three distinct scFvs that are specific to human SEMA3A using synthetic phage library.
3. Production of anti-SEMA3A IgG antibodies
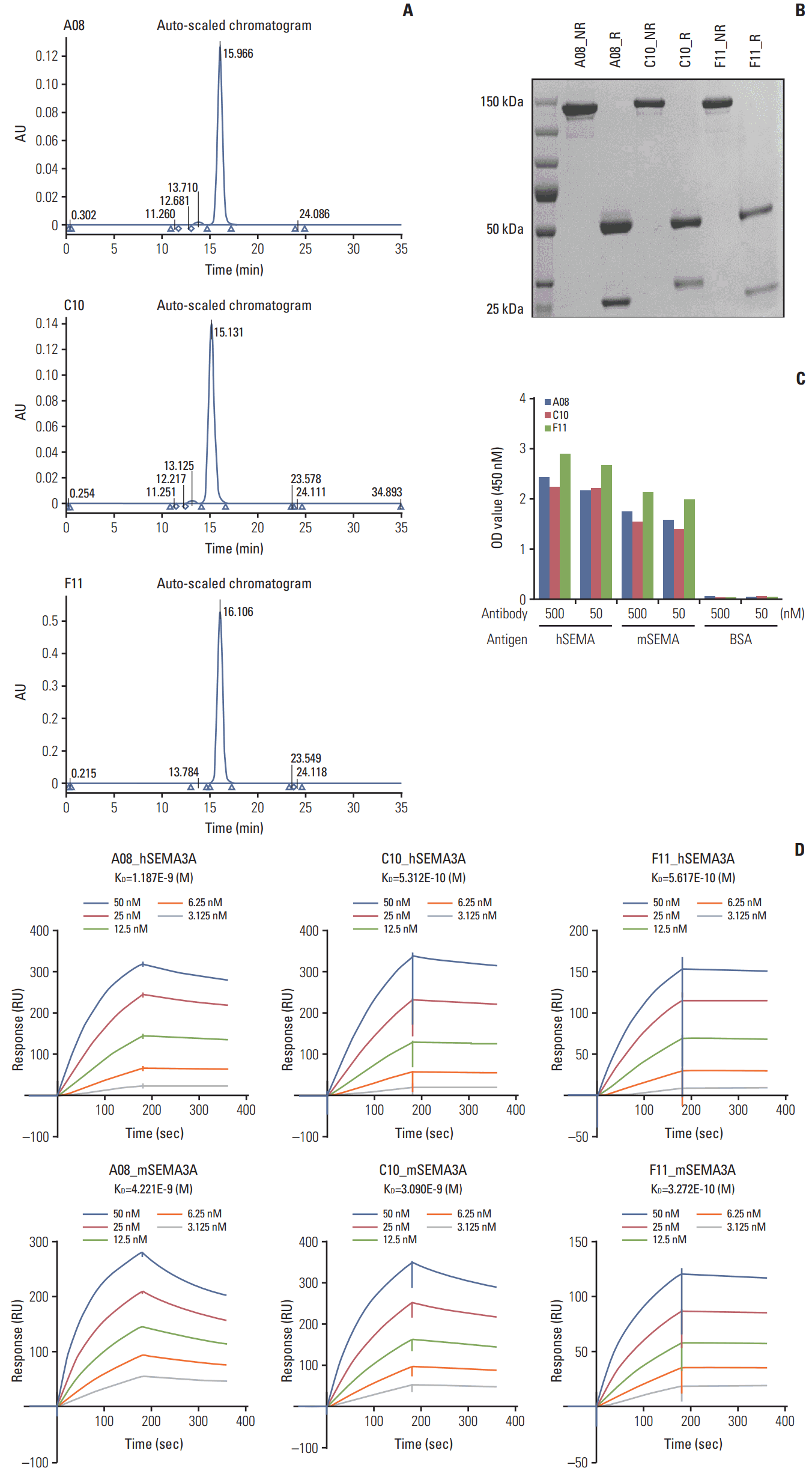
To transform the anti-SEMA3A scFvs into their respective IgG forms, we transfected Expi293F cells with heavy and light chain regions of each anti-SEMA3A scFv. We purified three anti-SEMA3A IgG antibodies using the ÄKTA Protein Purification System and an Amicon Ultra Centrifugal Filter to produce A08, C10, and F11 clones at concentration levels of 118, 138, and 330 mg/L, respectively. High performance liquid chromatography with a gel permeation chromatography column were used to validate the purity level of A08, C10, and F11 antibodies. Notably, all three clones exhibited 98%, 98.5%, and 99% purity, respectively (Fig. 3A). To validate the physical sizes of the anti-SEMA3A IgG antibodies, we conducted SDS-PAGE analysis under “reducing” and “non-reducing” conditions. Under the non-reducing condition, all three antibodies exhibited a molecular weight of 120 kDa, whereas, under the reducing condition, we observed two major bands at 50 and 25 kDa each, corresponding to the heavy and light chain regions of the antibody. Meanwhile, C10 contained N-glycosylation site in the CDR1 region of the light chain; thus, the molecular size of its light chain was at 30 kDa, 5 kDa higher than A08 and F11 (Fig. 3B).
To verify the binding activity of the anti-SEMA3A IgG antibodies to both human and mouse SEMA3A, we performed ELISA and surface plasmon resonance assays. As the mouse SEMA3A sequence harbors high homology to the human SEMA3A gene, at approximately 95%, all three antibodies showed positive cross-reactivity (Fig. 3C). Afterwards, we measured the KD (the equilibrium dissociation constant between the antibody and its antigen) values of all three SEMA3A antibodies and discovered that the F11 IgG antibody possessed the highest binding affinity capability to both human and mouse SEMA3A, consistent with the ELISA data (Fig. 3D). Collectively, our results demonstrate successful production of engineered anti-SEMA3A IgG antibodies that are specific to both human and mouse SEMA3A.
4. Anti-SEMA3A IgG antibody impedes migration and proliferation of patient-derived GBM cells
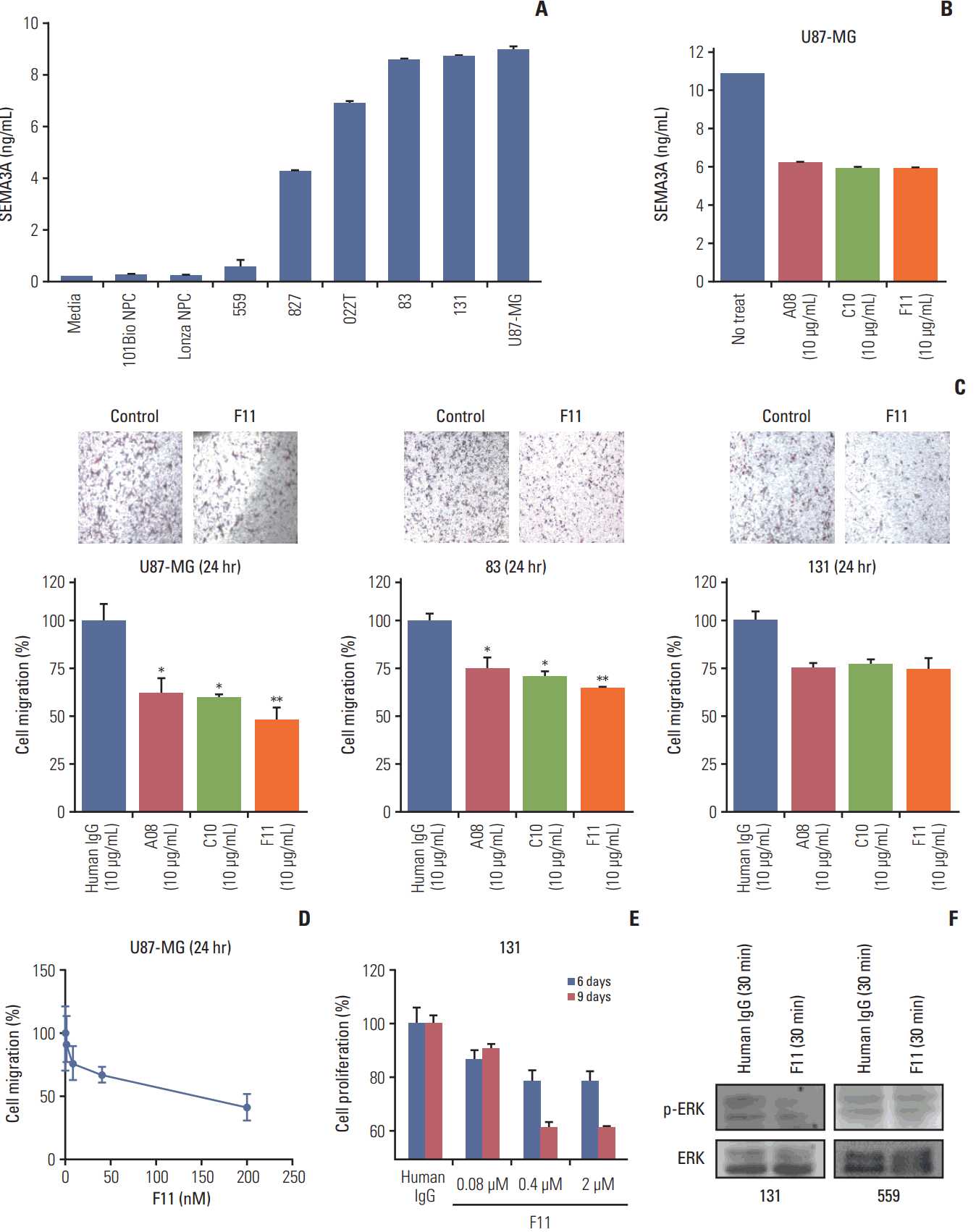
Previous reports have shown that GBM cells endogenously secrete SEMA3A and that RNA interference-mediated down regulation of SEMA3A was able to inhibit migratory ability of U87-MG cells [18]. To determine whether patient-derived GBM cells secret SEMA3A, we performed sandwich ELISAs with various PDCs, 131, 83, 022, 827, and 559, and U87-MG cell-line and neural progenitor cells (NPC) as positive and negative controls. Patient-derived GBM cells were cultured in NBA neurosphere culture conditions with additional growth factors (epidermal growth factor and basic fibroblast growth factor) and harvested and examined for secretion of SEMA3A. Both 131 and 83 cells secreted SEMA3A at a similar level to that of U87-MG cells, while the secretion level of SEMA3A was minimal in 559 tumor cells (Fig. 4A). Considering the neutralizing activity of our antibodies against SEMA3A, we performed a sandwich ELISA in the presence of anti-SEMA3A antibodies. Notably, all three antibodies were able to neutralize SEMA3A in U87-MG cells at 10 μg/mL.
Based on their SEMA3A neutralizing effects, we performed migration assays to determine the invasive inhibitory effect of anti-SEMA3A IgG antibodies. GBM cells were incubated in presence of different anti-SEMA3A IgG antibodies or control human IgG (10 μg/mL). Interestingly, all three antibodies showed significant migration suppression in U87-MG, 83, and 131 cells (Fig. 4C). As expected, the F11 antibody, which showed the highest KD value, exhibited greater inhibitory effects compared to the other antibodies. Using the Oris cell migration assay, we observed a dose-dependent migration inhibition of U87-MG cells in the presence of F11 antibody. (Fig. 4D). We then determined whether the proliferative response to anti-SEMA3A F11 was applicable in patient-derived GBM cells using Ez-Cytox cell viability assay. Under SEMA3A scFvs treatment, the cell proliferation rates were dependent on the secretion levels of SEMA3A from the GBM PDCs (S2 Fig.). Our results showed that the treatment with F11 antibody exhibited a dramatic reduction in cellular proliferation compared to the human IgG (Fig. 4E). Previous studies have shown that MAPK pathway including ERK1/2 induces cellular proliferation in gliomas [25]. Notably, immunoblot assay revealed that phospho-ERK level was significantly attenuated in 131 GBM cells that were pre-incubated with F11 for 30 minutes, whereas no notable changes were detected from the 559 cells (Fig. 4F). These results demonstrated that the F11 anti-SEMA3A IgG antibody effectively inhibited SEMA3A-induced migration and proliferation in GBM.
5. in vivo effects of anti-SEMA3A antibody on GBM tumor growth
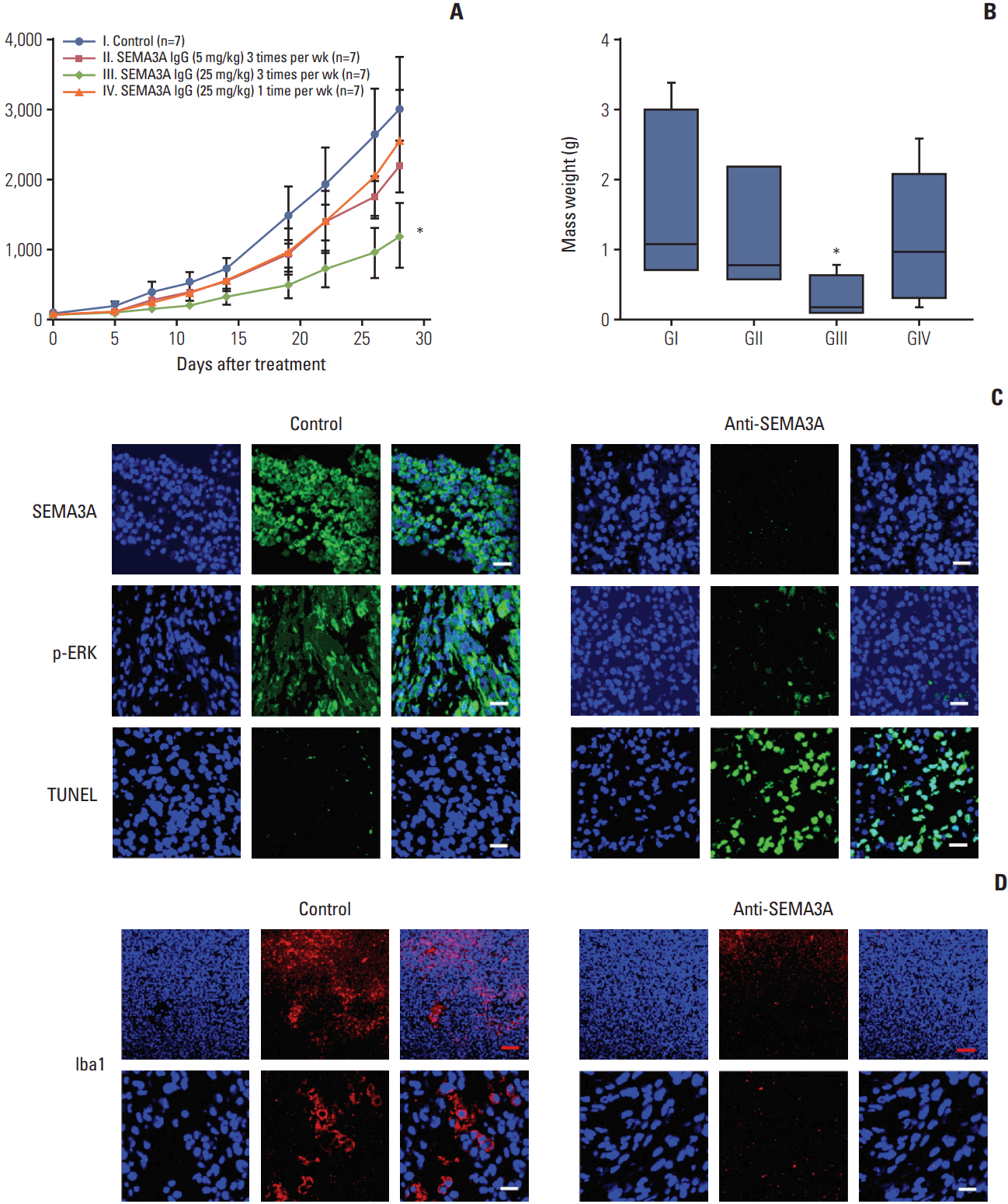
Based on in vitro migratory and proliferative inhibition effect of the F11 antibody, we further evaluated its efficacy in vivo setting. 131 tumor xenograft models were generated and treated with either PBS or F11 via intravenous injection. As suspected, treatment of tumor-bearing mice with 25 mg/kg of F11 antibody decreased the tumor growth by 60% compared to the PBS-treated group (Fig. 5A). Our results were consistently observed through tumor weight analysis in the corresponding mice (Fig. 5B). Mice that were treated with either F11 antibody or control showed no significant weight loss or neurological symptoms in S3 Fig.
Immunofluorescence staining of F11 antibody–treated tumors demonstrated significant reduction of SEMA3A and phosphor-ERK compared to the control tumor (Fig. 5C). Additionally, TUNEL assay showed F11 antibody-induced apoptotic cells, suggesting that F11 treatment can induce apoptotic cell death in vivo (Fig. 5C).
Previous studies have shown dynamic interaction between SEMA3A and tumor microenvironment. SEMA3A/NRP1 signaling promotes TAM infiltration and pro-tumorigenic function in hypoxic tumor regions [26,27]. Therefore, we interrogated the potential effect of SEMA3A neutralization on tumor microenvironment. Interestingly, TAMs were extensively recruited by adjacent tumor cells secreting SEMA3A in the control tumors, while significantly less accumulated TAM population was observed in the F11-treated tumors (Fig. 5D). Collectively, our results demonstrate that SEMA3A inhibitor could prevent tumor proliferation and TAM infiltration in vivo, highlighting anti-SEMA3A F11 antibody as a potential therapeutic agent in GBM therapy.
Discussion
SEMA3A is highly expressed in GBM compared to nonneoplastic brain tissues and SEMA3A signaling axis promotes GBM growth and invasion, making it an ideal therapeutic target in a clinical framework. Nevertheless, previous studies only showed RNA interference-mediated SEMA3A disruption that lead to migration inhibition [18]. Although siRNA has been widely used as an effective tool to down-regulate target genes, it cannot be translated into a clinical course. Therefore, we evaluated SEMA3A antibody as a potential therapeutic agent against GBM progression.
In present study, we have developed single-chain antibodies that are specific to Semaphorin3A from synthetic phage library using a classical panning method. To examine whether primary GBM cells secret SEMA3A, we used sandwich ELISA assay and compared the secretion level of SEMA3A in patient-derived GBM cells relative to NPC and U87-MG cells. We then converted the generated SEMA3A scFvs into three fully human IgG forms. IgG transformation not only increased their binding affinities, but also maintained their functional roles in cellular migration and proliferation inhibition. As the produced anti-SEMA3A antibodies are fully human, they may serve as a safer therapeutic agent in human disease. Furthermore, all three IgG antibodies showed specific cross-reactivity both mouse and human SEMA3A, allowing them to bypass in vivo immune response as well.
Previous studies have shown that SEMA3A modulates cell dispersal via activating multiple signaling pathways including Rac1, GSK3b, and ERK1/2 in various cancer cells. All three anti-SEMA3A antibodies were shown to repress migration of patient-derived GBM and U87-MG cell line. Furthermore, we confirmed the neutralization effect of anti-SEMA3A antibody using U87-MG cell line.
Dynamic interactions between tumor cells and tumor microenvironment frequently constitute tumor malignancy [28]. According to recent studies, TAMs are recruited to avascular areas in hypoxic tumor environment, resulting in sustained tumor progression [27]. SEMA3A acts as a chemoattractant by TAMs via VEGFR1 phophorylation and recruits TAMs towards hypoxic niches in NRP1-independent manner [26]. We confirmed that these results corroborate with our in vivo study demonstrating that anti-SEMA3A IgG suppresses tumor growth dramatically through preventing TAM recruitment.
Given that extracellular vesicle–derived SEMA3A as a propermeability factor that induces loss of barrier integrity in GBM, it could potentially be integrated for prognostic purposes. Our study also presents anti-SEMA3A IgG as a candidate diagnostic tool in monitoring GBM [19]. Recently, SEMA3A was evaluated as therapeutic target in ischemic or diabetic retinopathy, neurotrophic corneal disease, spinal cord injury and tumor progression [29]. Therefore, anti-SEMA3A antibody could be employed as a new diagnostic and therapeutic agent for GBM patients and other diseases that are related to SEMA3A.
 XML Download
XML Download