

 PDF
PDF Citation
Citation Print
Print


Introduction
Arising from the transformation of colon or rectum epithelial cells, colorectal cancer (CRC) is one of the most common malignancies worldwide and causes about 694,000 deaths every year [1]. During the initiation and progression of CRC, multiple germline and somatic mutations accumulated, particular for some critical oncogenes and tumor suppressors [2,3]. With great advances of the understanding of CRC pathogenesis, novel therapeutic strategies have been developed and greatly improved CRC patients’ prognosis, such as the molecularly targeted agents and immunotherapy [4]. However, the molecular mechanisms underlying CRC tumorigenesis are complex and many different driving events contribute to these processes [5,6]. Further revealing the underlying molecular mechanisms and developing more efficient therapeutic interventions could significantly improve the outcome of CRC patients.
Accumulating genomic and transcriptomic sequencing results have uncovered that only small proportion of the human genome is transcribed into protein-coding mRNAs, whereas the majority of the genome is transcribed into noncoding RNAs (ncRNAs) [7]. Among the ncRNAs, long noncoding RNA (lncRNA) is a class of novel identified ncRNAs with more than 200 nucleotides in length [8-10]. Recent evidences have revealed that lncRNAs have important roles in various pathophysiological processes, and are frequently deregulated in many diseases, particular in cancers [11,12]. lncRNA-HEIH has been reported to be upregulated in hepatocellular carcinoma (HCC) and facilitates HCC growth [13]. However, the clinical significances and roles of lncRNA-HEIH in CRC are still unknown.
MicroRNA (miRNA) is another class of functionally important ncRNAs with 21-25 nucleotides in length [14,15]. miRNAs have important roles in the regulation of gene expression via binding with mRNAs at complementary sites, and then inducing target mRNAs degradation and/or translational repression [16,17]. Except the well-known roles of miRNAs in mRNAs, several reports have shown that miRNAs directly modulate gene transcription via binding to promoters, enhancers, or transcription factors [18]. In our previous study, we have found that miR-939 physically binds to transcription factor nuclear factor κB (NF-κB), prevents the binding of NF-κB to the promoter of NF-κB target gene Bcl-xL, inhibits Bcl-xL transcription, and induces cell death [19]. Intriguingly, using bioinformatics analysis, we found three miR-939 binding sites on lncRNA-HEIH. Therefore, in this study, we further explored the interaction between lncRNA-HEIH, miR-939, NF-κB, and Bcl-xL. Moreover, we measured lncRNA-HEIH expression in CRC tissues and cell lines, analyzed its association with clinicopathological characteristics and prognoses of CRC patients. Using in vitro and in vivo functional assays, we investigated the roles of lncRNA-HEIH in CRC tumorigenesis. The contribution of the interaction between lncRNA-HEIH and miR-939 to the roles of lncRNA-HEIH were also detected.
Materials and Methods
1. Tissue specimens
A total of 84 paired CRC and adjacent normal mucosa tissues were obtained with informed consent from CRC patients who had received surgery at Zhujiang Hospital, Southern Medical University (Guangzhou, Guangdong, China). All the tissues were pathological diagnosed by the Department of Pathology, Zhujiang Hospital. The fresh tissue specimens were immediately frozen in liquid nitrogen until use.
2. Cell lines and cell culture
The human colon normal epithelial cell line NCM460 and CRC cell lines HT-29, SW480, HCT116, and LoVo were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). NCM460 was maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA). HT-29 and HCT116 were maintained in McCoy's 5A Medium (Sigma-Aldrich, St. Louis, MO). SW480 was maintained in L-15 Medium (Invitrogen). LoVo was maintained in Ham's F-12K Medium (Invitrogen). All the cells were grown in the above described medium supplemented with 10% fetal bovine serum (Invitrogen) at 37℃ in a humidified atmosphere with 5% CO2.
3. RNA extraction and quantitative real-time polymerase chain reaction
Total RNA was extracted from frozen tissues and culturing cells with the TRIzol reagent (Invitrogen) in accordance with the manufacturer’s protocol. Reverse transcription was performed with the M-MLV Reverse Transcriptase (Invitrogen) to produce the first strand cDNA. Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out with the SYBR Premix Ex Taq II (Takara, Dalian, China) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) in accordance with the manufacturer’s instructions. glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous control for lncRNA and mRNA. The primers used are as follows: for lncRNA-HEIH, 5'-CCTCTTGTGCCCCTTTCT-3' (forward) and 5'-AGGTCTCATGGCTTCTCG-3' (reverse); for Bcl-xL, 5'-CGTGGAAAGCGTAGACAAG-3' (forward) and 5'-AAGAGTGAGCCCAGCAGAA-3' (reverse); and for GAPDH, 5'-GGTCTCCTCTGACTTCAACA-3' (forward) and 5'-GTGAGGGTCTCTCTCTTCCT-3' (reverse). The expression of miR-939 and pri-miR-939 was measured by qRT-PCR as above described, using TaqMan MicroRNA Assay (Applied Biosystems) and TaqMan PrimiRNA Assay (Applied Biosystems) respectively, in accordance with the manufacturer’s protocols. The expression of target genes was calculated using the comparative Ct method.
4. Construction of vectors
Full length transcript of lncRNA-HEIH was polymerase chain reaction (PCR) amplified from cDNA using PrimeSTAR HS DNA polymerase (Takara) with the primers 5'-GGGGTACCGTCCCCGCCCCCTGC-3' (forward) and 5'-CGGGATCCCAAGGTTGGAAAATCCCACTTT-3' (reverse). The PCR amplified product was inserted into the Kpn I and BamH I sites of pcDNA3.1 (Invitrogen) and pSPT19 (Roche, Mannheim, Germany) vector, termed pcDNA3.1-HEIH and pSPT19-HEIH, respectively. The pcDNA3.1-HEIH and pSPT19-HEIH with mutations in miR-939 binding sites was synthesized by GenScript (Nanjing, China) and termed pcDNA3.1-HEIH-mut and pSPT19-HEIH-mut, respectively. Bcl-xL promoter region containing the NF-κB binding sites was PCR amplified from genomic DNA using PrimeSTAR HS DNA polymerase (Takara) with the primers 5'-GGGGTACCCAAGATCCACCACCCTCCAA-3' (forward) and 5'-CCCAAGCTTCCGTCTCTCCCGAACAAGC-3' (reverse), and inserted into the Kpn I and Hind III sites of the luciferase reporter pGL3-Basic (Promega, Madison, WI) vector, termed pGL3-Bcl-xL-promoter. Two independent shRNAs specifically targeting lncRNA-HEIH were designed as previously described [13], and then synthesized and inserted into the SuperSilencing shRNA expression vector pGPU6/Neo (GenePharma, Shanghai, China). The target sites of lncRNA-HEIH shRNAs were 5'-GCGCCUUCCCUCUAACCUUAA-3' and 5'-GGCAAGAUGAACGUCUGAAAU-3'. A scrambled shRNA was used as negative control for lncRNA-HEIH shRNAs.
5. Transfection of vectors and miR-939 inhibitors
miR-939 inhibitors and negative control were purchased from GenePharma. The transfections of vectors and miR-939 inhibitors were performed with the Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer’s protocol.
6. Construction of stable cell lines
For the construction of lncRNA-HEIH or lncRNA-HEIH-mut stably overexpressed HT-29 cells, pcDNA3.1-HEIH, pcDNA3.1-HEIH-mut, and pcDNA3.1 was transfected into HT-29 cells, and selected with neomycin (500 μg/mL) for four weeks. For the construction of lncRNA-HEIH stably silenced LoVo cells, the lncRNA-HEIH shRNAs and control shRNA were transfected into LoVo cells and selected with neomycin (500 μg/mL) for 4 weeks.
7. Cell proliferation assay
Cell proliferation was evaluated using the Cell Counting Kit-8 (CCK-8) assay and ethynyl deoxyuridine (EdU) incorporation assay. In brief, for CCK-8 assay, 2,000 indicated CRC cells were seeded in 96-well plates. The absorbance at 450 nm was detected with a CCK-8 (Dojindo Laboratories, Kumamoto, Japan) every 24 hours in accordance with the manufacturer’s instructions. EdU incorporation assay was carried out with an EdU Kit (Roche) in accordance with the manufacturer’s instructions.
8. Cell apoptosis assay
Cell apoptosis was evaluated using the TdT-mediated dUTP nick end labeling (TUNEL) assay with an In Situ Cell Death Detection Kit (Roche) in accordance with the manufacturer’s instructions. The apoptotic cells were labeled with green fluorescence, analyzed by fluorescence microscopy, and counted of at least five high-power fields.
9. in vivo tumor growth assay
In brief, 3×106 indicted CRC cells were subcutaneously injected into 5-weeks old female athymic BALB/c nude mice (Laboratory Animal Services Centre, Southern Medical University). Subcutaneous tumor growth was detected every 7 days with a caliper for 28 days. The tumor volume was calculated using the following formula: V=0.5×a×b2 (a, longitudinal diameter; b, latitudinal diameter). The nude mice experiments were performed in accordance with the guidelines for experimental animals of Zhujiang Hospital, Southern Medical University.
10. Immunohistochemistry and TUNEL staining
For immunohistochemistry staining of Ki-67, paraffin embedded tissue sections were deparaffinized, rehydrated, followed by antigen retrieval. After Ki-67 specific primary antibody (Cell Signaling Technology, Boston, MA) and a horseradish peroxidase-conjugated secondary antibody (Invitrogen) incubation, the proteins in situ were visualized with 3,3′-diaminobenzidine. For TUNEL staining, paraffin embedded tissue sections were dewaxed, rehydrated, protease treated, and permeabilized. Then TUNEL staining was performed using an In Situ Cell Death Detection Kit (Roche).
11. RNA pull-down assay
lncRNA-HEIH and miR-939 binding sites mutated lncRNA-HEIH (lncRNA-HEIH-mut) were in vitro transcribed from pSPT19-HEIH, and pSPT19-HEIH-mut, respectively, using the SP6 RNA polymerase (Roche) and biotinlabeled with the Biotin RNA Labeling Mix (Roche) in accordance with the manufacturer’s protocols. After being treated with RNase-free DNase I (Roche), the in vitro transcribed biotinylated RNA was purified using an RNeasy Mini Kit (Qiagen, Valencia, CA). Then 3 μg of purified biotinylated RNA was incubated with 1 mg of whole-cell lysates from HT-29 cells for 1 hour at 25℃, followed by being retrieved using the streptavidin agarose beads (Invitrogen). The RNA present in the pull-down material was quantified by qRT-PCR as previously described.
12. RNA immunoprecipitation assay
pcDNA3.1, pcDNA3.1-HEIH, or pcDNA3.1-HEIH-mut was transfected into HT-29 cells. lncRNA-HEIH shRNA or control shRNA was co-transfected with miR-939 inhibitors or negative control into LoVo cells. 48 hours after transfection, the cells were used to undergo RNA immunoprecipitation assay (RIP) assay with a NF-κB specific antibody (5 μg per reaction; Millipore, Bedford, MA) and the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) in accordance with the manufacturer’s protocol. The retrieved RNA was quantified by qRT-PCR as previously described.
13. Chromatin immunoprecipitation assay
pcDNA3.1, pcDNA3.1-HEIH, or pcDNA3.1-HEIH-mut was transfected into HT-29 cells. lncRNA-HEIH shRNA or control shRNA was co-transfected with miR-939 inhibitors or negative control into LoVo cells. Forty-eight hours after transfection, chromatin immunoprecipitation (ChIP) assay was carried out using a NF-κB specific antibody (5 μg per reaction, Millipore) and the EZ-Magna ChIP A/G Kit (Millipore) in accordance with the manufacturer’s protocol. The retrieved DNA was quantified by qRT-PCR as previously described with the primers 5'-CTCCTGTAAGCGAGTCTG-3' (forward) and 5'-TCGTTGAGCTATTGTGATG-3' (reverse), which spanning the NF-κB binding sites in Bcl-xL promoter region.
14. Luciferase reporter assay
pcDNA3.1, pcDNA3.1-HEIH, or pcDNA3.1-HEIH-mut was co-transfected with pGL3-Bcl-xL-promoter and pRL-TK (Renilla luciferase expression vector, Promega) into HT-29 cells. lncRNA-HEIH shRNA or control shRNA was co-transfected with miR-939 inhibitors or negative control, and pGL3-Bcl-xL-promoter and pRL-TK into LoVo cells. Forty-eight hours after transfection, luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega) in accordance with the manufacturer’s protocol. The relative firefly luciferase activity was normalized to Renilla luciferase activity.
15. Western blot
pcDNA3.1, pcDNA3.1-HEIH, or pcDNA3.1-HEIH-mut was transfected into HT-29 cells. lncRNA-HEIH shRNA or control shRNA was co-transfected with miR-939 inhibitors or negative control into LoVo cells. Forty-eight hours after transfection, the proteins were retrieved from these cells using RIPA buffer. Equal amounts of proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, followed by being blocked with 5% bovine serum albumin. Next, the membranes were incubated with primary antibodies specific for Bcl-xL (Abcam, Hong Kong, China) or GAPDH (Cell Signaling Technology), and IRDye 800CW goat anti-rabbit IgG or IRDye 700CW goat anti-mouse IgG (Li-Cor Biosciences, Lincoln, NE), and being detected with an Odyssey infrared scanner (Li-Cor Biosciences).
16. Statistical analysis
Results were analyzed using the GraphPad Prism Software (San Diego, CA). Statistical comparisons were subjected to Wilcoxon signed-rank test, Pearson chi-square test, log-rank test, Student’s t test, Mann-Whitney U test, or Pearson correlation analysis as indicated. p < 0.05 was considered as statistically significant.
Results
1. lncRNA-HEIH is increased in CRC tissues and cell lines, and correlated with poor outcome of CRC patients
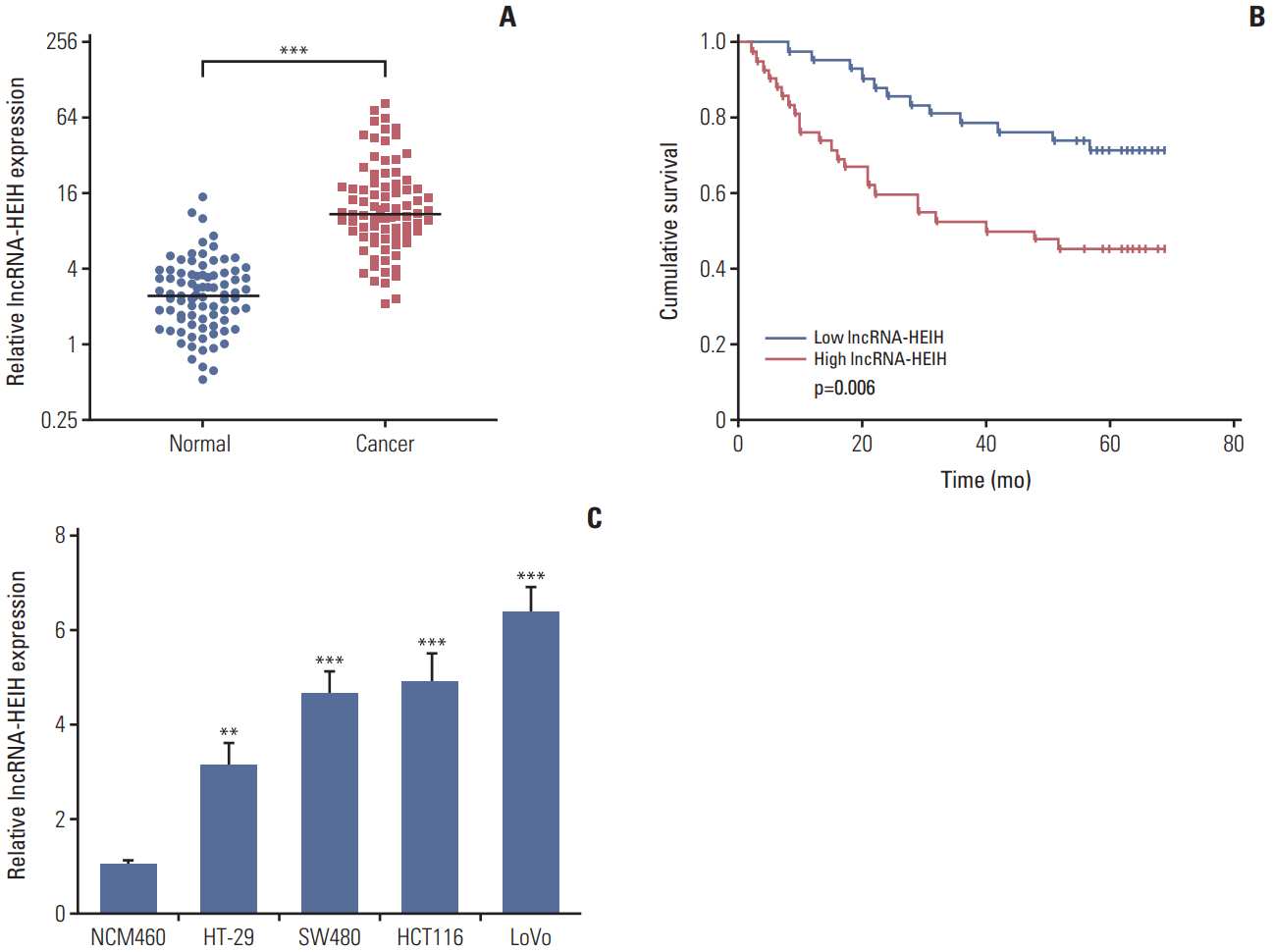
The expression of lncRNA-HEIH in 84 paired CRC and adjacent normal mucosa was detected using qRT-PCR. The result demonstrated that lncRNA-HEIH is significantly increased in CRC tissues compared with adjacent normal mucosa (Fig. 1A). Then, the correlation between lncRNA-HEIH expression and clinicopathological characteristics was analyzed in these CRC tissues. The results demonstrated that high expression of lncRNA-HEIH is associated with tumor size and invasion depth (T classification) (Table 1). A Kaplan-Meier survival analysis demonstrated that high expression of lncRNA-HEIH is correlated with poor overall survival (Fig. 1B). Multivariate Cox proportional hazards regression analysis further revealed that high expression of lncRNA-HEIH in CRC tissues was an independent prognostic factor for reduced overall survival (Table 2). Furthermore, the expression of lncRNA-HEIH in human colon normal epithelial cell line NCM460 and CRC cell lines HT-29, SW480, HCT116, and LoVo was also detected using qRT-PCR. The cell lines compared with human colon normal epithelial cell line (Fig. 1C). Taken together, these results demonstrated that lncRNA-HEIH is increased in CRC tissues and cell lines, and correlated with tumor size, invasion depth, and poor prognosis of CRC patients.
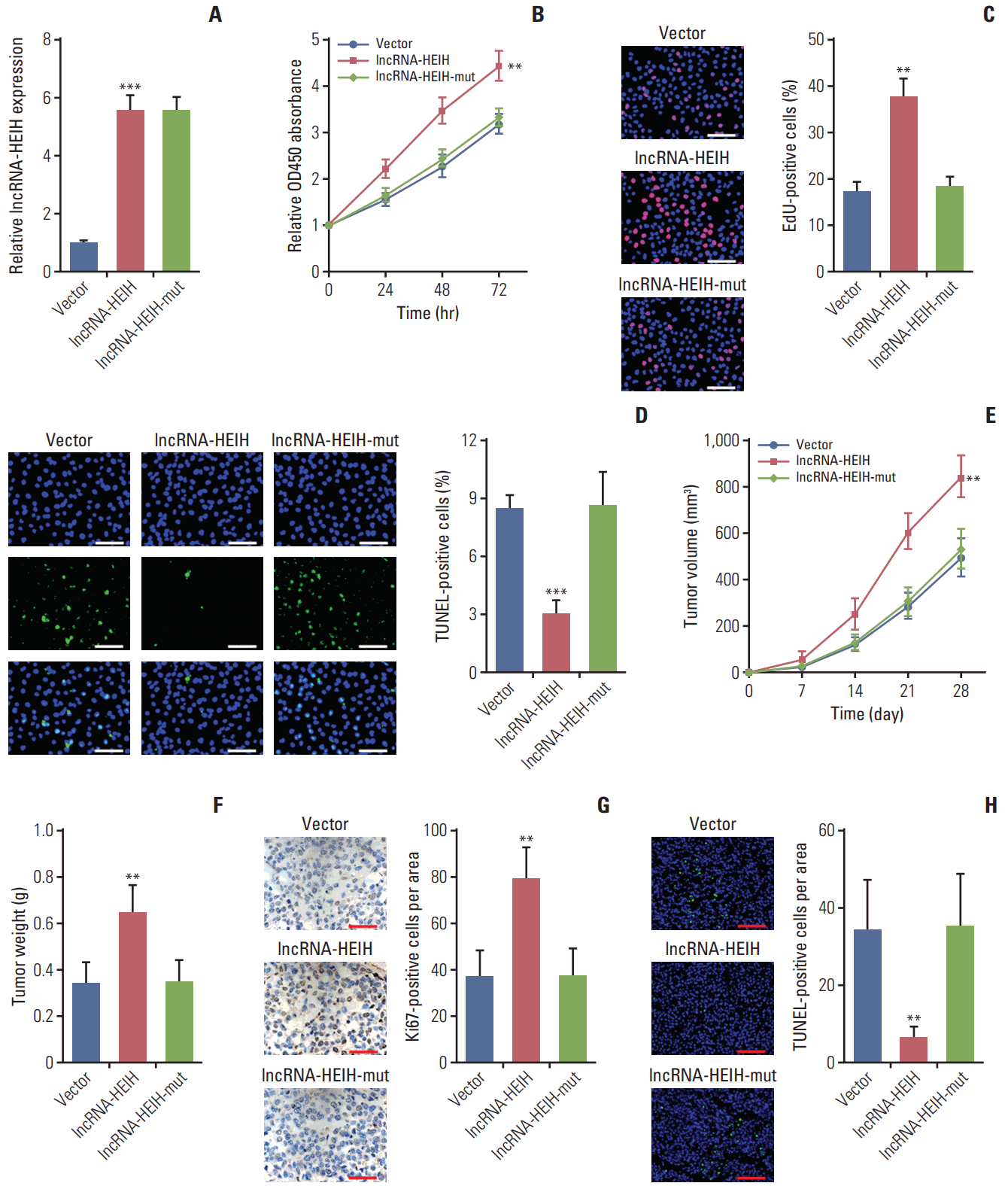
2. Enhanced expression of lncRNA-HEIH promotes CRC growth in vitro and in vivo
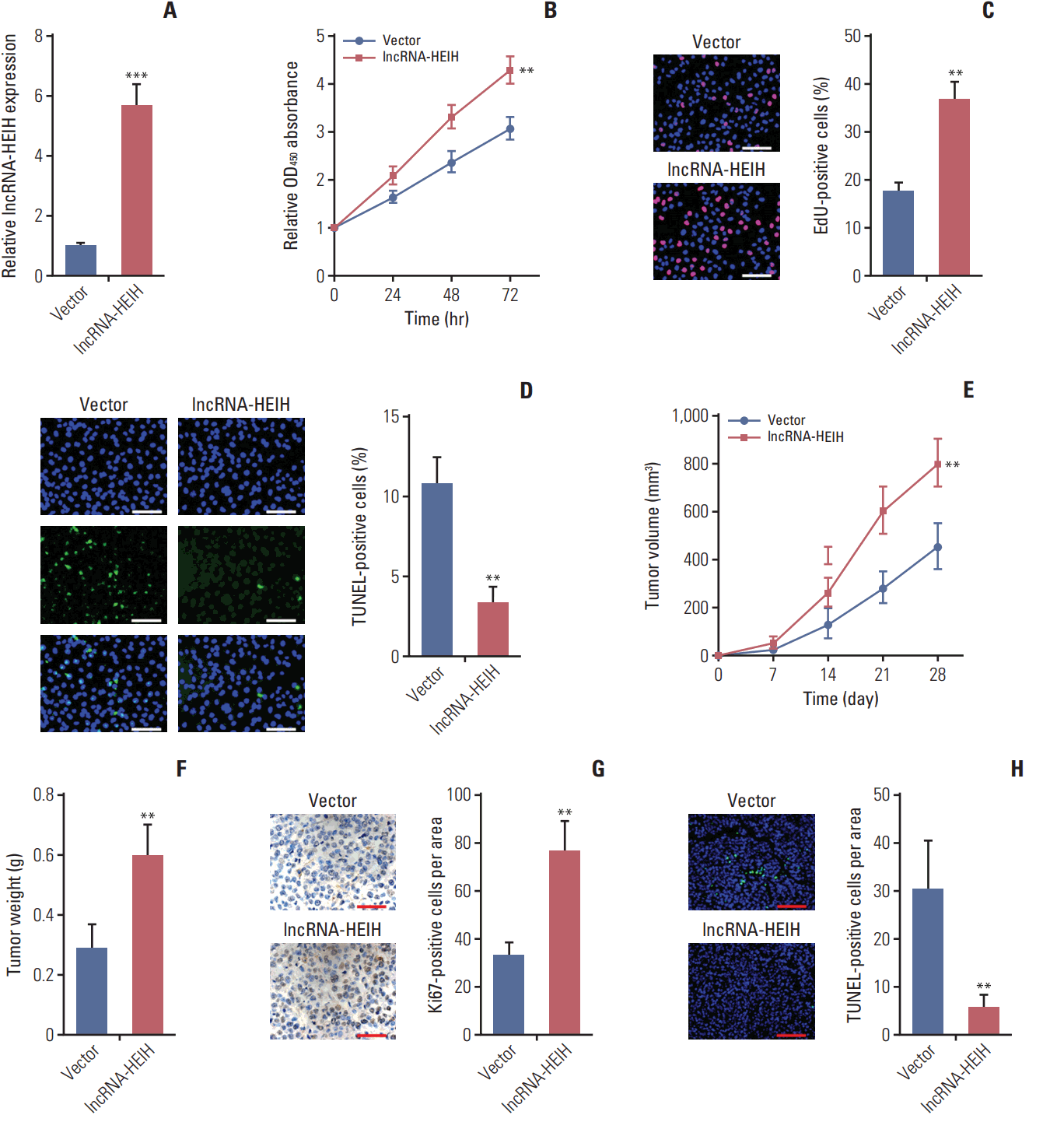
To determine the effects of lncRNA-HEIH on CRC cellular behavior, we transfected lncRNA-HEIH expression plasmid pcDNA3.1-HEIH into HT-29 cells and acquired lncRNA-HEIH stably overexpressed HT-29 cells (Fig. 2A). The CCK result demonstrated that lncRNA-HEIH is increased in CRC-8 assays demonstrated that enhanced expression of lncRNA-HEIH significantly increases the rate of cell proliferation in HT-29 cells (Fig. 2B). The EdU incorporation assays also demonstrated that overexpression of lncRNA-HEIH promotes HT-29 cell proliferation (Fig. 2C). The TUNEL staining demonstrated that enhanced expression of lncRNA-HEIH decreases the percentage of cells undergoing apoptosis in HT-29 cells (Fig. 2D). The effects of lncRNA-HEIH on CRC cellular behavior in vivo were also investigated. lncRNA-HEIH stably overexpressed and control HT-29 cells were subcutaneously injected into nude mice. The subcutaneous tumor model demonstrated that tumors grew faster in HT-29/lncRNA-HEIH overexpression group compared with HT-29/control group (Fig. 2E and F). Immunohistochemistry staining of proliferation marker Ki-67 demonstrated that enhanced expression of lncRNA-HEIH increases the number of cells undergoing proliferation in tumors formed from HT-29 cells (Fig. 2G). The TUNEL staining of subcutaneous tumors demonstrated that enhanced expression of lncRNA-HEIH decreases the number of cells undergoing apoptosis (Fig. 2H). Taken together, these results demonstrated that enhanced expression of lncRNA-HEIH promotes CRC growth in vitro and in vivo.
3. Knockdown of lncRNA-HEIH inhibits CRC growth in vitro and in vivo
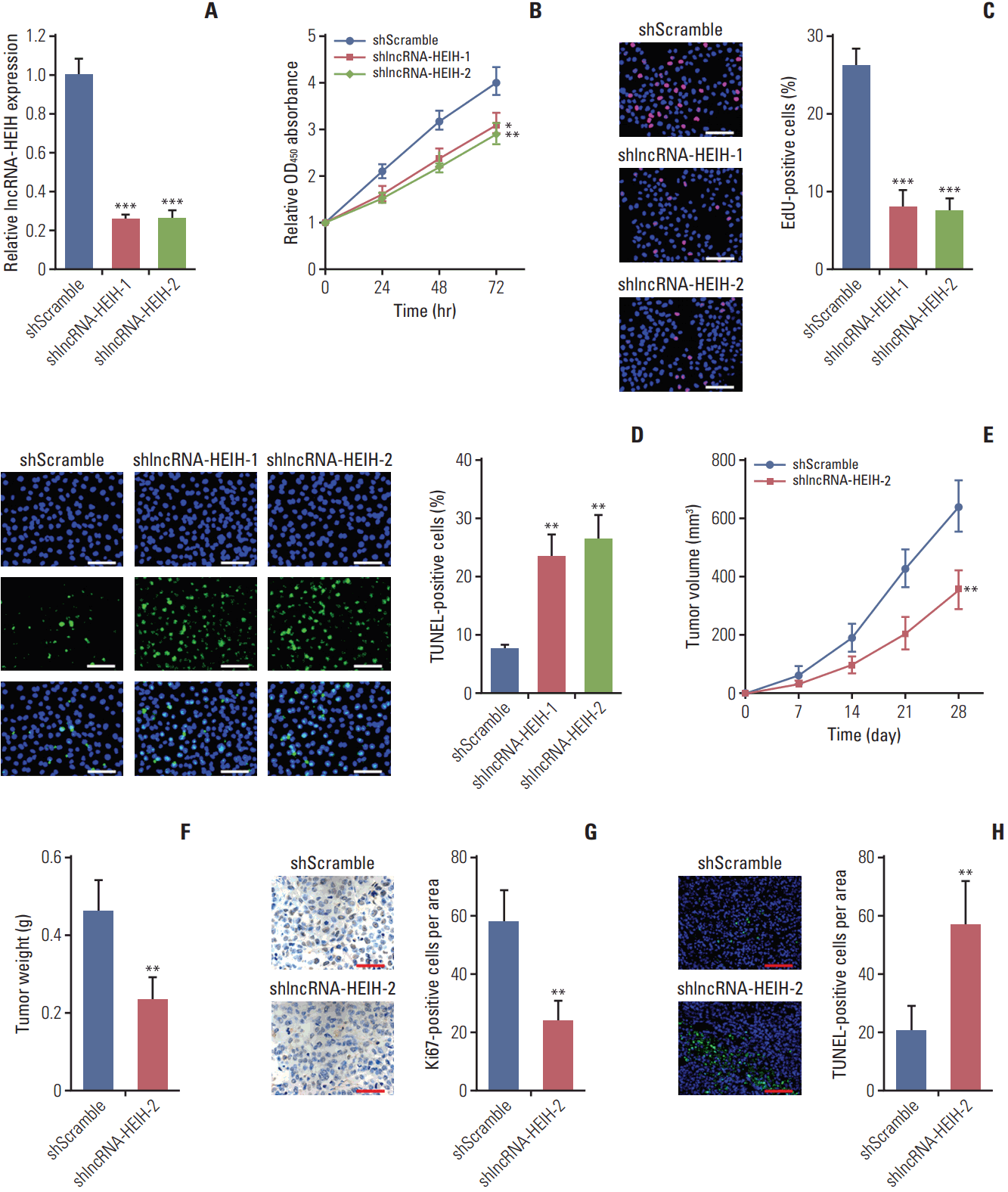
To completely investigate the effects of lncRNA-HEIH on CRC cellular behavior, we transfected two independent lncRNA-HEIH shRNAs into LoVo cells and acquired lncRNA-HEIH stably knocked-down LoVo cells (Fig. 3A). The CCK-8 assays demonstrated that knockdown of lncRNA-HEIH by both shRNAs decreases the rate of cell proliferation in LoVo cells (Fig. 3B). The EdU incorporation assays also demonstrated that knockdown of lncRNA-HEIH by both shRNAs inhibits LoVo cell proliferation (Fig. 3C). The TUNEL staining demonstrated that knockdown of lncRNA-HEIH increases the percentage of cells undergoing apoptosis in LoVo cells (Fig. 3D). The effects of lncRNA-HEIH knockdown on CRC cellular behavior in vivo were further investigated. lncRNA-HEIH stably silenced and control LoVo cells were subcutaneously injected into nude mice. The subcutaneous tumor model demonstrated that tumors grew more slowly in LoVo/lncRNA-HEIH knockdown group compared with LoVo/control group (Fig. 3E and F). Immunohistochemistry staining of Ki-67 demonstrated that knockdown of lncRNA-HEIH decreases the number of cells undergoing proliferation in tumors formed from LoVo cells (Fig. 3G). The TUNEL staining of subcutaneous tumors demonstrated that knockdown of lncRNA-HEIH increases the number of cells undergoing apoptosis (Fig. 3H). Taken together, these results demonstrated that knockdown of lncRNA-HEIH inhibits CRC growth in vitro and in vivo.
4. lncRNA-HEIH physically associates with miR-939 and suppresses the binding between miR-939 and NF-κB
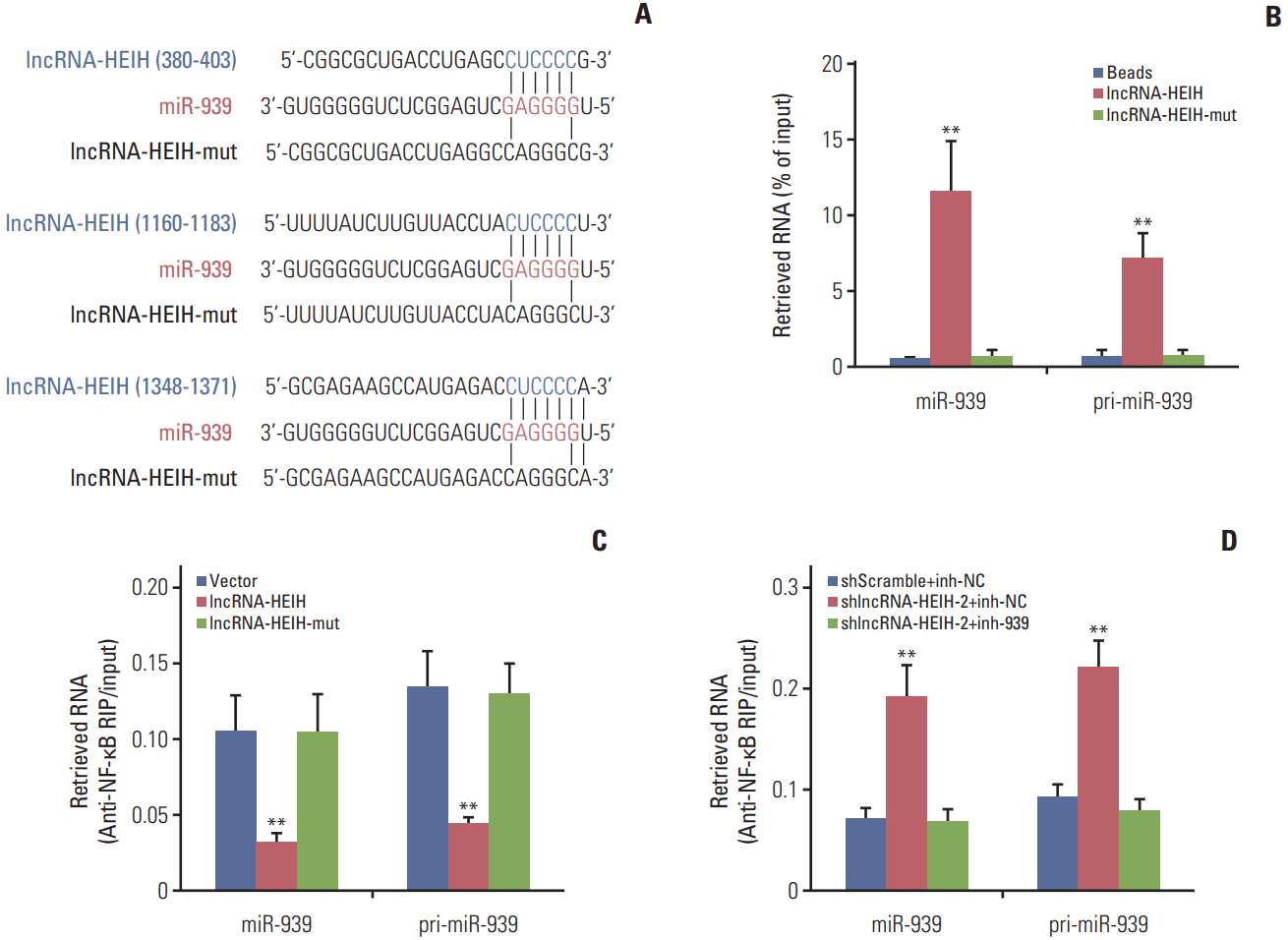
To explore the molecular mechanisms underlying the effects of lncRNA-HEIH on CRC cellular behavior, we performed bioinformatics analysis by TargetScan and found three putative miR-939 response elements on lncRNA-HEIH (Fig. 4A). To investigate whether lncRNA-HEIH could physically associates with miR-939, we carried out RNA pulldown assays using in vitro transcribed biotinylated lncRNA-HEIH. The RNA pull-down assays demonstrated that lncRNA-HEIH specifically binds to miR-939 and the primary miR-939 (pri-miR-939), and while mutation of the miR-939 response elements on lncRNA-HEIH abolishes the binding of lncRNA-HEIH to miR-939 and pri-miR-939 (Fig. 4B). In our previous study, we found that miR-939 could physically bind to transcription factor NF-κB and produce transcriptional regulation of NF-κB target gene Bcl-xL. Next, we investigated whether the association between lncRNA-HEIH and miR-939 influences the binding of miR-939 to NF-κB. RIP assays using NF-κB specific antibody demonstrated that enhanced expression of lncRNA-HEIH significantly decreases the binding of NF-κB to miR-939 and pri-miR-939, and while mutation of the miR-939 response elements on lncRNA-HEIH abolishes the effects of lncRNA-HEIH on the binding of NF-κB to miR-939 and pri-miR-939 (Fig. 4C). The RIP assays also demonstrated that knockdown of lncRNA-HEIH significantly increases the binding of NF-κB to miR-939 and pri-miR-939, and while inhibition of miR-939 reverses the effects of lncRNA-HEIH knockdown on the binding of NF-κB to miR-939 and pri-miR-939 (Fig. 4D). Taken together, these results demonstrated that lncRNA-HEIH physically associates with miR-939 and suppresses the binding between miR-939 and NF-κB.
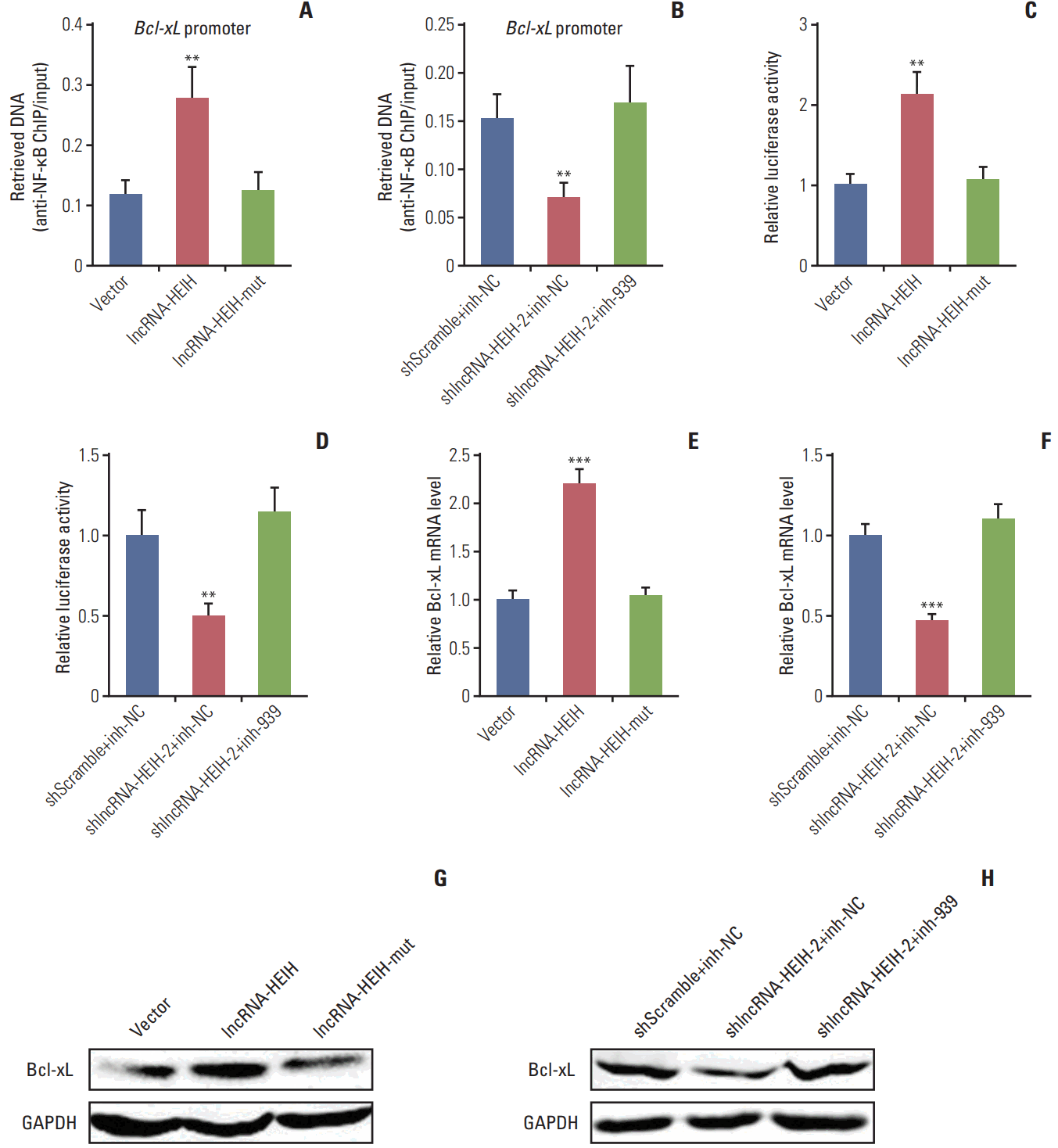
5. lncRNA-HEIH counteracts miR-939-mediated transcriptional repression of Bcl-xL
In our previous study, we have found that through binding to NF-κB, miR-939 decreases the occupancy of NF-κB on the promoter region of the Bcl-xL gene and transcriptionally represses Bcl-xL expression [19]. Bcl-xL is a NF-κB target and has critical anti-apoptotic roles in many cancers [20]. Next, we investigated the effects of lncRNA-HEIH on the transcriptional regulation of Bcl-xL by miR-939. The ChIP assays using NF-κB specific antibody demonstrated that enhanced expression of lncRNA-HEIH significantly increases the binding of NF-κB to Bcl-xL promoter region, and while mutation of the miR-939 response elements on lncRNA-HEIH abolishes the effects of lncRNA-HEIH on the binding of NF-κB to Bcl-xL promoter (Fig. 5A). The ChIP assays also demonstrated that knockdown of lncRNA-HEIH significantly decreases the binding of NF-κB to Bcl-xL promoter region, and while inhibition of miR-939 reverses the effects of lncRNA-HEIH knockdown on the binding of NF-κB to BclxL promoter (Fig. 5B). These data suggested that through competitively binding to miR-939, lncRNA-HEIH promotes the binding of NF-κB to Bcl-xL promoter. Then, the luciferase reporter assays were performed to investigate the effects of lncRNA-HEIH on transcription activity of Bcl-xL. Bcl-xL promoter region spanning the NF-κB binding sites was inserted into the luciferase vector. The luciferase reporter assays demonstrated that enhanced expression of lncRNA-HEIH significantly increases the luciferase activities of the construct containing Bcl-xL promoter, and while mutation of the miR-939 response elements on lncRNA-HEIH abolishes the increasing of the luciferase activities (Fig. 5C). The luciferase reporter assays also demonstrated that knockdown of lncRNA-HEIH significantly decreases the luciferase activities of the construct containing Bcl-xL promoter, and while inhibition of miR-939 reverses the decreasing of the luciferase activities (Fig. 5D). These data suggested that through competitively binding to miR-939, lncRNA-HEIH increases the transcription activity of Bcl-xL.
The effects of lncRNA-HEIH on the expression of Bcl-xL were further detected by qRT-PCR and western blot assays. The qRT-PCR assays demonstrated that enhanced expression of lncRNA-HEIH significantly increases Bcl-xL mRNA levels, which was abolished by the mutation of the miR-939 response elements on lncRNA-HEIH (Fig. 5E). Knockdown of lncRNA-HEIH significantly decreases Bcl-xL mRNA levels, which was abolished by miR-939 inhibition (Fig. 5F). Western blot assays demonstrated that enhanced expression of lncRNA-HEIH significantly increases Bcl-xL protein levels, which was abolished by the mutation of the miR-939 response elements on lncRNA-HEIH (Fig. 5G). Knockdown of lncRNA-HEIH significantly decreases Bcl-xL protein levels, which was abolished by miR-939 inhibition (Fig. 5H). Taken together, these results demonstrated that lncRNA-HEIH increases the transcription activity and expression of Bcl-xL via competitively binding to miR-939 and suppressing the binding between miR-939 and NF-κB.
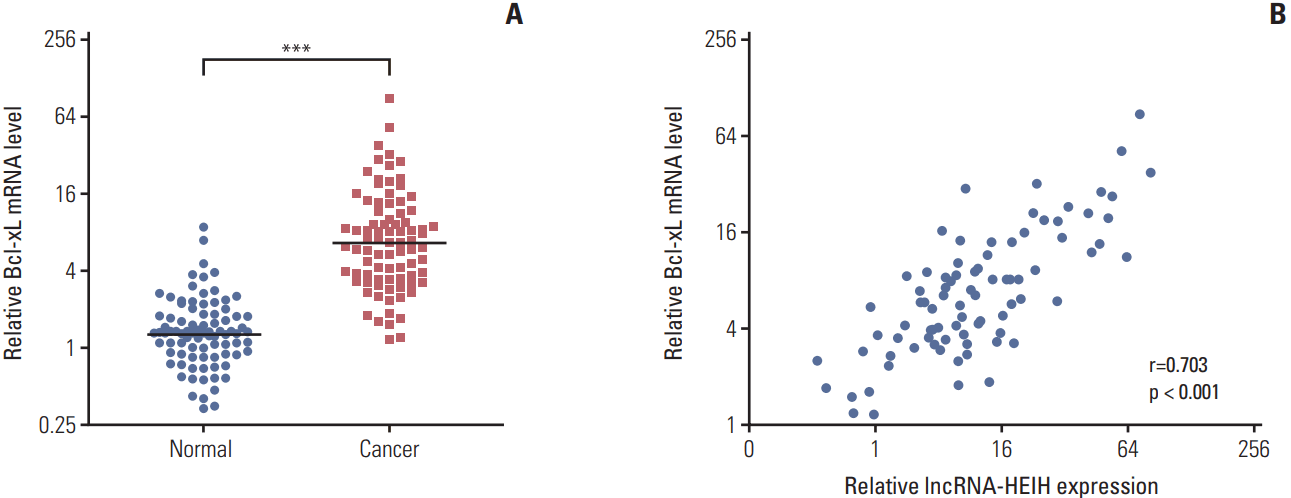
6. Bcl-xL expression is positively correlated with lncRNA-HEIH expression in CRC tissues
The expression of Bcl-xL in the same 84 paired CRC and adjacent normal mucosa was detected using qRT-PCR to determine whether Bcl-xL expression is associated with lncRNA-HEIH expression in clinical tissue samples. The result demonstrated that Bcl-xL is also significantly increased in CRC tissues compared with adjacent normal mucosa (Fig. 6A). A correlation analysis demonstrated that Bcl-xL expression level is positively correlated with that of lncRNA-HEIH in CRC tissues (Fig. 6B).
7. The effects of lncRNA-HEIH on CRC tumorigenesis are dependent on the binding to miR-939
To determine whether the effects of lncRNA-HEIH on CRC tumorigenesis are dependent on the competitive binding to miR-939, we transfected miR-939 response elements mutated lncRNA-HEIH expression plasmid into HT-29 cells and acquired lncRNA-HEIH-mut stably overexpressed HT-29 cells with the same overexpression efficiency as lncRNA-HEIH stably overexpressed HT-29 cells (Fig. 7A). The CCK-8 assays demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the increasing of the cell proliferation rate caused by lncRNA-HEIH overexpression in HT-29 cells (Fig. 7B). The EdU incorporation assays also demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the pro-proliferative effects of lncRNA-HEIH in HT-29 cells (Fig. 7C). The TUNEL staining demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the decreasing of the percentage of cells undergoing apoptosis caused by lncRNA-HEIH overexpression in HT-29 cells (Fig. 7D). The subcutaneous tumor model demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the pro-growth effects of lncRNA-HEIH (Fig. 7E and F). Immunohistochemistry staining of proliferation marker Ki-67 demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the increasing of the number of cells undergoing proliferation in tumors formed from HT-29 cells caused by lncRNA-HEIH overexpression (Fig. 7G). The TUNEL staining of subcutaneous tumors demonstrated that the mutation of the miR-939 response elements on lncRNA-HEIH abolishes the decreasing of the number of cells undergoing apoptosis caused by lncRNA-HEIH overexpression (Fig. 7H). Taken together, these results demonstrated that the effects of lncRNA-HEIH on CRC tumorigenesis are dependent on the competitively binding to miR-939.
Discussion
CRC is the third most common and one of the most frequent cause of cancer-related death worldwide [1]. Increased cell survival capability is critical during CRC tumorigenesis and progression [21]. Previously, people mainly focused their attentions on proteins with oncogenic or tumor-suppressing roles, such as KRAS, p53, and adenomatous polyposis coli (APC) [22]. Recently, tens of thousands of noncoding transcripts have been identified [23]. Many of these non-coding transcripts showed attractive important roles in cancers, which provides new opportunities for the diagnosis and therapy of cancers [24]. A recently identified lncRNA lncRNA-HEIH localizes at chromosome 5q35.3, is polyadenylated, and has 1,681 nucleotides in length [13]. In this study, we further explored the expression, clinical significances, and roles of lncRNA-HEIH in CRC. We found that lncRNA-HEIH is significantly up-regulated in CRC tissues and cell lines, positively associated with tumor size and invasion depth, and indicts poor prognosis of CRC patients. in vitro and in vivo functional experiments demonstrated that enhanced expression of lncRNA-HEIH promotes CRC cell proliferation, decreases CRC cell apoptosis, and promotes CRC tumor growth in vivo. Whereas knockdown of lncRNA-HEIH inhibits CRC cell proliferation, induces CRC cell apoptosis, and suppresses CRC tumor growth in vivo. These data suggest that up-regulated lncRNA-HEIH functions as an oncogene in CRC, and imply that lncRNA-HEIH may be a potential prognostic biomarker and therapeutic target for CRC.
The action mechanisms of lncRNAs are complex. lncRNAs could physically bind to DNA, proteins, mRNAs, or miRNAs, and change the expression, localization, and function of the binding partners [11]. Among the diverse action mechanisms, the competitive bindings to miRNAs of lncRNAs are increasingly reported [8]. Through competitively binding common miRNAs, lncRNAs relieve the degradation and/or translation repression of targeted mRNAs caused by the common miRNAs. However, accumulating evidences reveal that, except the roles of miRNAs in the cytoplasm, miRNAs are also found in the nucleus [25]. These nuclear miRNAs could activate or repress gene transcription via binding to promoters, enhancers or transcription factors. In our previous study, we have revealed that miR-939 physically binds to NF-κB, exerts decoy-like actions to prevent the binding between NF-κB and the promoter of its target gene Bcl-xL, and further represses Bcl-xL transcription [19]. Interestingly, we identified three miR-939 binding sites on lncRNA-HEIH. RNA pull-down assays confirmed the interaction between lncRNA-HEIH and miR-939. RIP and ChIP assays demonstrated that the interaction between lncRNA-HEIH and miR-939 damages the binding between miR-939 and NF-κB, counteracts the effects of miR-939 on the binding between NF-κB and Bcl-xL promoter. Luciferase reporter assays, qRT-PCR, and western blot showed that the interaction between lncRNA-HEIH and miR-939 counteracts the effects of miR-939 on the transcription and expression of Bcl-xL. Furthermore, the expression of Bcl-xL is positively associated with that of lncRNA-HEIH in CRC tissues. Blocking the interaction between lncRNA-HEIH and miR-939 by the mutation of miR-939 binding sites on lncRNA-HEIH abolishes the biological effects of lncRNA-HEIH on CRC tumorigenesis. These data suggest that lncRNA-HEIH promotes CRC tumorigenesis through counteracting the roles of miR-939. Except for Bcl-xL, miR-939 has been reported to regulate the expression of SLC34A2, APC2, and VE-cadherin in other cancers [26]. Through binding to miR-939, whether lncRNA-HEIH also regulates the expression of these miR-939 targets in CRC needs further investigation. Metastasis is the common reason of CRC patients’ death. Whether lncRNA-HEIH regulates the proliferation and/or apoptosis of metastatic CRC cells in a similar manner also needs further investigation. Furthermore, whether there is correlation between lncRNA-HEIH and well-known genes of colon cancer carcinogenesis such as KRAS, p53, and APC is not clear and also needs further investigation.
In conclusion, our study showed that lncRNA-HEIH is up-regulated in CRC tissues and cell lines, associated with tumor size, invasion depth, and poor outcome of CRC patients, promotes CRC cell proliferation and inhibits CRC cell apoptosis in vitro, and promotes CRC tumor growth in vivo via counteracting miR-939-mediated transcriptional repression of Bcl-xL. Our results suggested that lncRNA-HEIH may be a novel diagnostic biomarker and therapeutic target for CRC.
 XML Download
XML Download