

 PDF
PDF Citation
Citation Print
Print


Introduction
Lung cancer is an important public health problem and the leading cause of cancer death worldwide [1]. The two major histological types of lung cancer are non-small cell lung cancers (NSCLC) and small cell lung cancers. NSCLCs consist mostly of lung adenocarcinomas (ADCA) and lung squamous cell carcinomas (SQCC). These two NSCLC subtypes have both unique and common clinical and histopathological characteristics. For example, ADCAs are mainly observed in never smokers [2] while SQCC is the most frequent histological type among smoking patients.
The discovery of common, mutually exclusive, driver genes, epidermal growth factor receptor (EGFR), KRAS proto-oncogene GTPase (KRAS) and anaplastic lymphoma receptor tyrosine kinase (ALK), that represent crucial therapeutic targets, led to a remarkable improvement in NSCLC therapy. However, only about 50% of NSCLC acquire mutations in these genes. Identification of new cancer driver genes currently is a top research priority. Recent studies using nextgeneration sequencing (NGS) approach characterized NSCLC tissues in order to identify new less common candidate driver mutations [3,4] highlighting the presence of great variability even within tumor samples belonging to the same histological type.
Up to now, application of NGS technology has been reported in many lung cancer samples leading to the discovery of a large set of new somatic mutations in key cancer genes. Nevertheless, no studies are present in literature that performed whole-exome sequencing (WES) and RNA-sequencing (RNA-seq) from tumoral and matched nontumoral lung tissues integrating these with WES of blood DNA from the same individual to identify possible tumordriver variants which are already present in the germline background and that may predispose to lung cancer. Using WES analysis in a model of cases and controls deriving from the same kindred—discordant sib-pair design—we have recently showed that a set of oligogenic germline mutations, that are detectable in the peripheral blood, may play a role in the development and growth of lung ADCA in never smokers [5]. Noteworthy, our previous results demonstrated that the oligogenic cancer predisposing signature is a “private signature,” varying from one patient to another.
Herein, we tested further the hypothesis of the oligogenic origin of lung cancer in a never-smoker female who developed a SQCC of the lung. We applied for the first time an integrative “omic” approach of WES and RNA-seq experiments in a discordant sib-pair design.
Materials and Methods
1. Samples and DNA/RNA extraction
Patient and sib signed the informed consent declaration at the Medical Genetics Unit of the Azienda Ospedaliera Universitaria Senese, Italy, for the use of their biological samples and clinical data for research purposes. The study protocol was approved by the Ethical Committee of the Azienda Ospedaliera Universitaria Senese. Information on histological diagnosis (by the Pathology Unit) was retrieved from the clinical records. Genomic DNA of patient and healthy sib was isolated from EDTA peripheral blood using QIAamp DNA Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Formalin-fixed paraffin-embedded (FFPE) samples of patient’s tumoral and non-tumoral lung tissues were obtained from the Pathology Unit of the recruiting hospital and analyzed through WES and RNA-seq. DNA was extracted from FFPE lung tumoral and non-tumoral tissue samples using MagCore Genomic DNA FFPE One-Step Kit for MagCore System (Diatech Pharmacogenetics s.r.l., Ancona, Italy). RNA was extracted from FFPE lung tumoral and non-tumoral tissue samples of our case using High Pure FFPE-Tissue RNA Isolation Kit (Roche, Basel, Switzerland) following the manufacturer’s instructions. Additional RNAs from three FFPE lung non-tumoral tissues from other lung cancer patients were isolated using the same kit and used as control tissues. RNA samples were processed to remove rRNA using Ribo-Zero rRNA Removal Kit for Human samples (Illumina, Grand Island, NY) following the manufacturer’s instructions. RNA integrity was verified using the Agilent Eukaryote Total RNA Nano Kit (Agilent Technologies, Palo Alto, CA) on Agilent2100 Bioanalyzer (Agilent Technologies). Both DNA and RNA were quantified by spectrophotometry (ND-2000c, NanoDrop Products, Wilmington, DE) and Qubit Fluorometer with Qubit dsDNA HS Assay and Qubit RNA HS Assay Kits (Life Technologies, Carlsbad, CA), respectively.
2. WES and data analysis
WES was performed using the Life Technologies Ion Proton sequencer (Life Technologies) on genomic DNA samples of cases and controls and tumor tissues as previously described [6]. Variant were then annotated against external datasets, including 1000 genomes (http://www.1000genomes.org/) and dbSNP. After removing variants with low coverage, in order to identify candidate susceptibility variants, we selected for rare variants with minor allele frequency [MAF] ≤ 0.01 or not reported (S1 Fig.). We then filtered excluding variants with clinical significance as “benign” or “likely benign” and present in an in-house database of variants. Additional filtering procedures were thus implemented for retrieving exonic rare variants with a potential detrimental impact on protein function, i.e., variants, insertions and deletions (indels) causing exonic frameshifts and missense variants predicted as deleterious applying the Combined Annotation Dependent Depletion (CADD > 25) and the MetaSVM (Support Vector Machine) bioinformatics tools. Genes affected by mutations were checked in literature in order to identify cancer-related genes. Validation of these variants was carried out using custom NGS panel for Ion PGM sequencer (Life Technologies).
3. RNA-seq and data analysis
RNA-seq was performed using Illumina HiSeq2500 platform (Illumina Inc., San Diego, CA), in a 2×100 bp pairedend configuration in High Output mode (V4 chemistry), with a total of at least 250 million reads per lane. After quality check, RNA (50 ng) was used to prepare sample libraries. Sequencing library construction included these steps: library construction using Illumina TruSeq RNA Sample Pre Kit (Illumina), library purification using Beckman AMPure XP beads (Beckman Coulter s.r.l., Milan, Italy), insert fragments test using Agilent High Sensitivity DNA Kit on Agilent 2100 Bioanalyzer (Agilent Technologies), and cBOT automatic cluster (TruSeq PE Cluster Kit v3-cBotHS). Post-library quality controls were performed using the Agilent RNA 6000 Nano kit (Agilent Technologies) on Agilent 2100 Bioanalyzer (Agilent Technologies) and Qubit Fluorometer (Life Technologies). Libraries were then loaded on HiSeq2500 sequencing platform (Illumina) and sequenced using 2×100 bp pair-end High Output Mode (V4 chemistry) per lane. The reads generated on the HiSeq2500 were provided under FASTQ format.
Sequence reads in FASTQ format were processing using the Fastqc software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) for data quality check and removing excess adaptors to get high-quality and clean reads. The high-quality reads were aligned to the GRCh38/hg38 reference human genome (ftp://jgi-psf.org/pub/compgen/phytozome/v9.0/Ptrichocarpa/assembly/Ptrichocarpa_210.fa.gz) using the TopHat software ver. 2.0.9 [7]. Transcript assembling and expression quantification were carried out using Cufflinks ver. 2 [8]. Gene expression was expressed as fragments per kilo-base transcript per million mapped reads (FPKM) value. This normalized value was used for visualization on a genome browser (http://genome.ucsc.edu/), as well as to compare read coverage between and throughout different genes. Statistical analysis was performed to compute the mean FPKM level with the associated p-value for lung normal tissues together with the mean FPKM level with the associated p-value for lung cancer tissues. Cuffdiff tool from Cufflinks was used to identify differentially expressed genes [9]. Gene Ontology and pathways analyses were performed using both Enrichr (http://amp.pharm.mssm.edu/Enrichr/) and Database for Annotation, Visualization and Integrated Discovery functional annotation tool (DAVID Bioinformatics Resources 6.7, https://david.ncifcrf.gov).
Results
1. Patient description
A still alive Caucasian female patient, who developed SQCC of the lung in the absence of smoking habit and/or asbestos exposure, was recruited. Her smoker father died at age 63 from lung cancer. She had no known other family history of cancers. At age of 77, after a “screening” chest and abdomen computed tomography scan examination, a 12×7 mm nodule was found in the dorsal segment of the right superior lobe in association with a small “ground glass” area (9.5 mm in diameter) in the left superior lobe. The nodule showed a small volumetric increase after 3 months and the patient was scheduled for surgery at the Thoracic Surgery Unit of Azienda Ospedaliera Universitaria Senese, Italy. Intraoperative diagnosis was NSCLC. At gross pathologic examination, a 12×0.5 solid nodule with irregular margin was detected. Histologic examination showed SQCC with a mainly perialveolar and perivascular growth pattern. Tumoral cells were p63+, cytokeratin (CK) 5/6+, CK7‒, thyroid transcription factor 1‒, synaptophysin‒, chromogranin‒. Ki67 immunostaining was 20 per cent. There was moderate infiltration of the visceral pleura, but no lymphonodal infiltration. Two years before, patient had undergone to removal of a cutaneous nodule located in the left side of the neck that resulted to be a SQCC of the basal subtype. She subsequently underwent left cervical lymphadenectomy, that showed infiltration of three cervical lymphonodes. On the basis of histological analysis, site of tumors and type of lymphonodal involvement, it was stated that the patient had two different primary tumors, both belonging to the squamous subtype. The patient is alive and well 4 years after the first and 2 years after the second operation.
2. “Omic” characterization
We firstly carried out WES of gDNA from FFPE tumoral and non-tumoral lung tissue. With the aim of identification of somatic driver mutations, we subtracted the germline background to 39,286 variants present in the tumoral lung tissue, leading to 2,543 exonic variants present only in the tumor. Among them, 1,381 were missense variants and 254 were truncating (S2A Fig.). We then extracted the somatic mutational signature according to base substitutions, as described by Alexandrov et al. (2013) [10]. This analysis showed a predominance of C>T transitions in tumoral lung tissue, not corresponding to the specific cancer signature related to tobacco consumption that are characterized by predominance of C>A transversions (S2B Fig.). Pathway analysis showed that mutations observed in SQCC tissue mostly affected genes involved in extracellular matrix organization (p=0.005), transmembrane transport of small molecules (p=0.010) and collagen formation (p=0.034) pathway, as previously reported (S2C Fig.) [4]. Overall, no mutations in the known driver genes, such as EGFR, KRAS, AKT, ROS1, nor in other putative driver genes described by Campbell et al. [3] were present in our tumoral sample [3]. EML4-ALK fusion gene was also excluded. The failure of identification of somatic driver mutations rose the hypothesis that germline variants may have a role in etiology of the disease.
We then adopted a novel integrative “omic” approach using a combination of NGS techniques in a pedigree model that compares discordant sibs to test the hypothesis of oligogenic origin of lung cancer (Fig. 1).
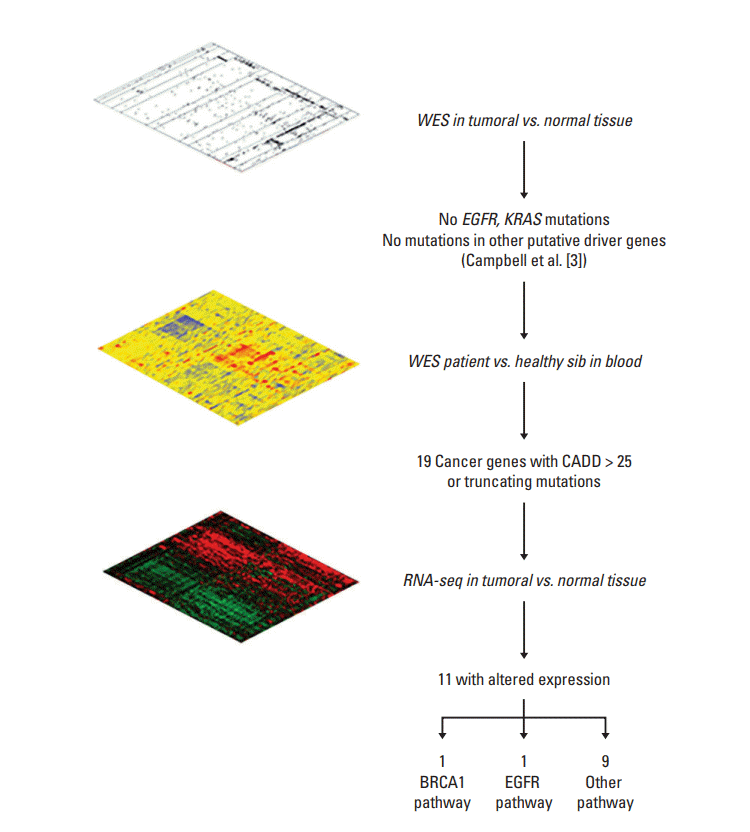
We carried out WES of gDNA from peripheral blood of the SQCC patient and her unaffected sib as controls (S3 Table). Overall, we identified 37,727 germline variants in the patient and 37,355 germline variants in the healthy sib and 39,286 variants in the tumoral lung tissue.
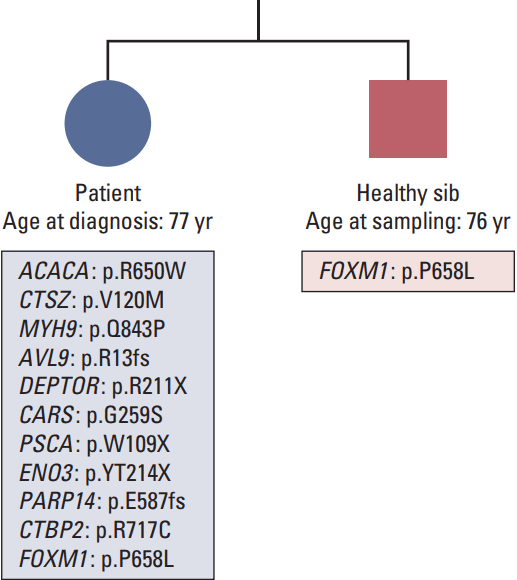
To identify susceptibility variants, we compared case and control WES data and selected for rare variants (MAF ≤ 0.01) with a potentially damaging effect: truncating variants, i.e., insertions and deletions (indels) causing exonic frameshifts and nonsense mutation, and missense deleterious variants predicted with CADD > 25 and filtering out those with MetaSVM as “tolerated” (S1 Fig.). We obtained 111 potential deleterious variants of which 40 were shared by both siblings. Among the 111 potentially deleterious variants, 19 variants mapped in genes that have been found previously associated with cancer of the lung or other tissues (S4 Table). Validation of these variants led to the confirmation of all variants that were probably responsible for lung cancer susceptibility in our case. All the sequence variants identified were in the heterozygous state and were also present in the relative lung tumor tissue in heterozygous state as expected.
Finally, we performed RNA-seq analysis of RNA extracted from both FFPE tumoral and non-tumoral lung tissues of our patient in order to detect differences in the expression level that could help the understanding of the functional role of germline variants. RNA-seq generated a mean of 75,134,860 reads per sample (S5 Table).
To assess the potential functional role of the 19 germline cancer-related variants identified by WES in the patient and predicted as deleterious by bioinformatics tools, we combined results from WES experiments with the respective-gene expression profile from RNA-seq of nontumoral and matched tumoral tissue. In 19 variants mapping in 19 genes, RNA-seq data showed differential expression level (fold-change > ±1.5) for 11 genes, reinforcing the pathogenic role of the identified variants showing three different effects (Table 1). Three genes, namely ACACA, CTSZ, and MYH9, showed downregulation in lung cancer compared to normal tissue. Three genes, namely DEPTOR, AVL9, and CARS, showed downregulation of both normal and cancer tissue when we compared those tissues with a pool of three normal lung tissues. Lastly, five remaining genes (PSCA, ENO3, PARP14, CTBP2, FOXM1) showed an upregulation in lung cancer tissue. All the above mentioned variants were exclusive of the patient with the exception of variant in FOXM1 gene that was present also in the healthy sib.
Table 1.
Expression levels of 11 cancer-related germline variants (fold-change > ±1.5)
| Group | Gene symbol | Amino acid change (CADD value) | Fold-change T/N | Gene function related to cancer development |
|---|---|---|---|---|
| Group 1: Underexpressed in tumoral tissue (fold-change ≤ 0.66) | ACACA | p.R650W (33) | –4.35 | Acetyl-CoA carboxylase alpha has a role in de novo lipogenesis catalyzing the carboxylation of acetyl-CoA to malonyl-CoA. The enzyme is under long term control at the transcriptional and translational levels. It is known to specifically interact with BRCA1 [11]. |
| MYH9 | p.Q843P (31) | –1.64 | Myosin heavy chain 9 is a myosin IIA heavy chain that interacts with β-actin to maintain cytoskeleton integrity. It is a EGFR-associating protein in NSCLC cell lines overexpressing EGFR [12]. | |
| CTSZ | p.V120M (26) | –1.59 | Cathepsin Z is a lysosomal cysteine proteinase. CTSZ is mostly expressed in immune cells, such as macrophages and monocytes, and involved in antigen processing and maturation of the major histocompatibility complex class II molecules [13]. This gene is expressed ubiquitously in cancer cell lines and primary tumors. | |
| Group 2: Underexpressed in both normal and tumoral tissue (fold-change ≤ 0.66) | AVL9 | p.R13fs (NA) | –3.13a) | AVL9 cell migration associated is among the 73 cancer driver candidate genes previously identified via dog-human comparison, deleted in colorectal cancer [14] and classified as putative tumor suppressors. AVL9-depletion inhibits the migration of A549 human adenocarcinomic alveolar basal epithelial cells [15]. |
| CARS | p.G259S (33)b) | –3.03a) | Cysteinyl-tRNA synthetase is one of several located near the imprinted gene domain on chromosome 11p15.5, an important tumor-suppressor gene region [16]. | |
| DEPTOR | p.R211X (46) | –1.96a) | DEP-domain containing mTOR interacting protein has growth suppression activity against pancreatic cancer cells, and its expression is gradually lost during pancreatic tumorigenesis [17]. Tumor progression mediated by EGFR ectopic expression was diminuished by transfection with DEPTOR in lung adenocarcinoma. Gefitinib, a specific EGFR inhibitor used for lung cancer therapy, stimulated DEPTOR accumulation [18]. | |
| Group 3: Overexpressed in tumoral tissue (fold-change ≥ +1.5) | PSCA | p.W109X (NA)b) | 391.96 | Prostate stem cell antigen encodes a glycosylphosphatidylinositol-anchored cell membrane glycoprotein that is upregulated in a large proportion of prostate cancers and other cancers. It is highly expressed in non-small cell lung cancer and may be functionally important for the disease: small interfering RNA-mediated knockdown of PSCA resulted in the inhibition of LC growth [19]. |
| ENO3 | p.YT214X (35) | 7.04 | Enolase 3 encodes one of the three enolase isoenzymes. It was found upregulated in liver cancer mouse model [20]. | |
| PARP14 | p.E587fs (NA) | 4.93 | Poly(ADP-ribose) polymerase family member 14 is an anti-apoptotic protein that may regulate aerobic glycolysis and promote survival of cancer cells [21]. Its increased expression has been reported in a variety of tumor types. | |
| CTBP2 | p.R717C (34) | 4.34 | C-terminal binding protein 2 is upregulated in a number of tumors, such as hepatocellular carcinoma, prostate cancer, and gliomas [22, 23]. | |
| FOXM1 | p.P658L (27)b) | 3.28 | Forkhead box M1 is a transcriptional activator involved in cell proliferation. Consistent with an important role of Foxm1 in cell cycle progression, increased expression of Foxm1 was found in many human tumors. Increased Foxm1 expression in human lung adenocarcinomas and squamous cell carcinomas was associated with increased proliferation of tumor cells [24]. |
CADD, Combined Annotation Dependent Depletion; T, tumoral lung tissue; N, non-tumoral lung tissue; ACACA, acetyl-CoA carboxylase alpha; EGFR, epidermal growth factor receptor; NSCLC, non-small cell lung cancers; mTOR, mammalian target of rapamycin; NA, not applicable; LC, lung cancer; MAF, minor allele frequency.
![]()
Among the 11 germline-mutated cancer-related genes, two genes, namely ACACA and DEPTOR, are known to be associated with the known driver genes, BRCA1 and EGFR, respectively. In particular, we found a missense variant, that has been predicted as deleterious, c.C1948T, p.Arg650Trp (NM_198837.1, exon 16) in ACACA gene (OMIM* 200350) encoding Acetyl-CoA carboxylase alpha, the rate-limiting enzyme for the long-chain fatty acid synthesis [25]. The other mutation is a stopgain variant c.A631T, p.Arg211* in DEPTOR gene (OMIM *612974) encoding DEP domain-containing MTOR-interacting protein.
Interestingly, from RNA-seq data, we found a decreased expression level of ACACA in tumoral rather that in nontumoral tissue. Sequence information was extracted from RNA-seq to confirm the presence of mutated mRNA and we found that 81.8% of reads carried the mutated allele pinpointing to a loss of heterozygosity (LOH) effect. The same analysis revealed a decreased expression level of DEPTOR both in normal and tumoral tissue at mRNA level pinpointing to LOH effect already present in the surrounding normal tissue.
Discussion
Among 111 potentially deleterious germline variants, 19 mapped in genes that have previously been associated with cancer of the lung or other tissues (S3 Table). Using the discordant- sib pair design, we confirmed the presence of an oligogenic signature present in the patient’s genetic background [5]. In fact, only one out of 11 cancer-related variants was also found in the healthy sib. For all the 11 variants, RNA-seq data reinforced the pathogenic role showing three different effects on the expression levels of harboring gene: three genes (ACACA, CTSZ, MYH9) showed a possible “second hit” in tumor suppressor genes responsible for gene inactivation; three genes (AVL9, DEPTOR, CARS) showed a possible transcript instability occurring in both tumoral and normal mutation-bearing tissues; five genes (PSCA, ENO3, PARP14, CTBP2, FOXM1) were upregulated in lung cancer tissue confirming previous published results and suggesting their role as oncogene. No mutations or gene expression alteration in genes belonging to the Hedgehog pathway, mainly involved in basal cell carcinoma development, were found in our case.
The oligogenic combination of cancer predisposing genes found in our patient supports the model of “private genetic epidemiology” for lung cancer susceptibility [5]. In fact, we found that 11 cancer-related genes were mutated in the patient and only one in healthy sib (Fig. 2), suggesting that the oligogenic control of lung cancer susceptibility is due to a peculiar signature of multiple germline mutations in cancer-related genes probably with a dosage effect. This personal signature may play a role in the development and growth of lung cancer and, therefore explain the non-hereditability of the condition, especially in non-smokers patients.
Focusing our analysis on genes that have already been reported in association with known druggable target genes—and likely having a major role in driving tumor development in our case—, we identified two possible suitable targets. Acetyl-CoA carboxylase alpha (ACACA) is a key enzyme in de novo lipogenesis catalyzing the carboxylation of acetyl-CoA to malonyl-CoA. ACACA is known to be one of the major BRCA1-interacting protein [11]. Cancer cells strongly need to de novo synthesize fatty acid for membrane and energy production [26] and interaction between ACACA and BRCA1 is likely to be important for BRCA1-mediated tumor suppressor activity [27]. BRCA1 indeed binds the phosphorylated and inactive form of ACACA and this interaction blocks ACACA activity by preventing its dephosphorylation [27]. The identified variant c.C1948T, p.Arg650Trp is outside the catalytic domain but may have a role in folding of domain conformation determining a different exposure for BRCA1 binding. Based on the evidence that in our tumoral tissue only the mutated transcript is present, we can assume that, even if protein level is decreased, the isoform loses negative control by BRCA1 and remains active. In this case, patient could potentially benefit from a therapy with ACACA inhibitors such as miR-195 [28] or soraphen A [29] (Fig. 3A).
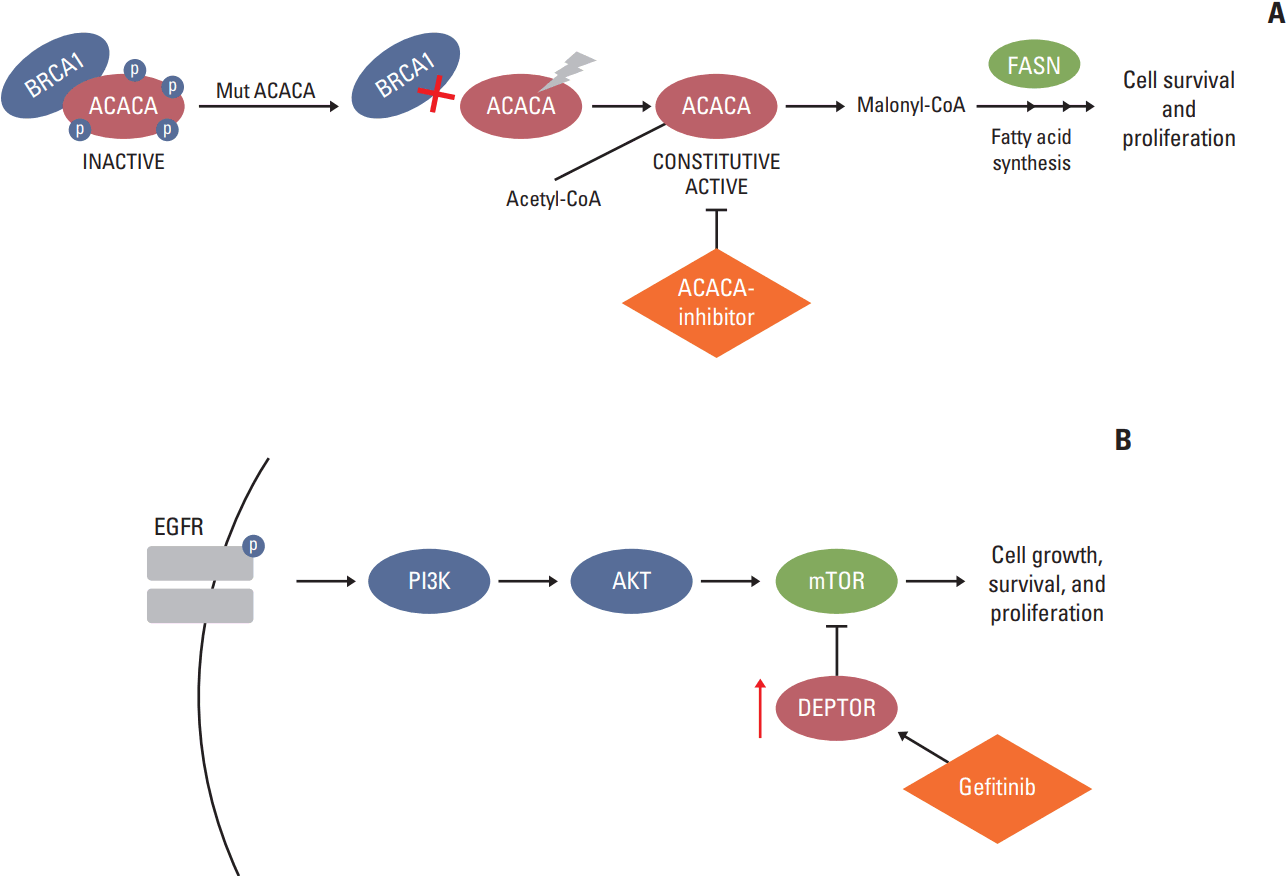
The second druggable variant maps in DEPTOR gene, whose loss determines the activation of mammalian target of rapamycin (mTOR) cascade and promotes cell growth and survival. It is known that DEPTOR mainly acts as a tumor suppressor [17,30]. A recent study in lung cancer demonstrated that activation of EGFR signaling resulted in downregulation of DEPTOR expression. In the same study, tumor progression mediated by EGFR ectopic expression was diminished by transfection with DEPTOR in lung adenocarcinoma cell lines and that treatment with gefitinib, an EGFR tyrosine kinase inhibitor, stimulated DEPTOR expression leading to a significantly decreased cell proliferation, migration and invasion [18]. In line with these results, we observed an increased expression level of DEPTOR in normal and tumoral tissue, suggesting an enhanced predisposition of activation of mTOR signaling and thus a promotion of tumor progression (Fig. 3B). These results suggested the possibility to use gefitinib in our patient in order to restore DEPTOR level and consequently suppress cell growth and survival.
In summary, we suggest that both “primary” SQCC are triggered by the presence of an oligogenic germline signature consisting of at least 11 mutations, among which two lead to the activation of mTOR and BRCA1. Thus, this finding opens up the possibility of treating one of them with an already commercially available drug and the other with developing molecules.
This study has a number of limitations. First, alterations in non-coding regions and copy number variants would have been missed by our approach. Another limitation is that we focused only on cancer-associated genes, but we cannot exclude that non-cancer genes, in particular those involved in immune system or drug metabolism, could play a role in cancer development. Finally, additional functional studies will be required to translate our results into effective personalized therapy.
The present report provides evidence that an “omic” sequencing approach in cancer patient is feasible, and could impact on treatment decisions, namely in patients not suitable for conventional therapy. In the present era of “precision medicine”, our study demonstrates the utility of applying this comprehensive sequencing protocol in order to effectively identify possible therapeutic targets enabling personalized therapies.
 XML Download
XML Download