

 PDF
PDF Citation
Citation Print
Print


Introduction
Gastric cancer (GC) constitutes a major cause of cancer-related deaths worldwide, but its management, especially in advanced stages, has evolved relatively little. Human epidermal growth factor receptor 2 (HER2/ErbB2/neu) is a 185-kDa transmembrane receptor tyrosine kinase (RTK) and a member of the epidermal growth factor receptor (EGFR) family [1]. HER2 serves as an important therapeutic target for therapy in HER2-positive metastatic GC since its overexpression is found in more than 15% of GC and is associated with poor prognosis, particularly in the advanced stages of disease [2]. For patients with HER2-positive advanced GC, trastuzumab (Herceptin, Genentech) combined with standard chemotherapy has been used as first-line treatment [2,3]. However, intrinsic and/or acquired resistance totrastuzumab became a major obstacle in anti-HER2 therapy for advanced GC [2]. Thus, there is a need for alternatives to block HER2 signaling in GC.
Lapatinib (Tykerb, GlaxoSmithKline, Ware, UK) is an oral dual tyrosine kinase inhibitor of EGFR (HER1) and HER2 [4]. Although lapatinib could be used for treatment of trastuzumab-resistant, HER2-positive advanced GC cases, the major problem of therapies targeting tyrosine kinases is that many tumors do not response or eventually develop resistance to the drugs, often due to activation of alternative signaling pathways [5]. Therefore, it is important to know in advance which pathways could mediate resistance to the lapatinib treatment and to find ways of bypassing these obstacles [6].
Mesenchymal-epithelial transition factor protein (MET), the hepatocyte growth factor (HGF) receptor, is a 190-kDa RTK, and plays a critical role in tumor growth, invasion and metastasis. MET is frequently overexpressed and activated in a subset of GC [7]. Previously it has been shown that co-expression of MET and HER2 in GC is associated with poorer survival compared to overexpression of either one [8]. Moreover, MET overexpression occurred more frequently in HER2-positive GCs than in HER2-negative GCs [9]. Growing evidences implicate the interplays between HER family receptors and MET in cancer cells through overlapping downstream signaling pathways [6]. in vitro cell culture experiments showed that HGF-induced MET activation was responsible for lapatinib resistance in HER2-positive GC cell lines [10,11]. In addition, GC cells derived from HER2-positive and MET-positive GC showed that the combination of lapatinib and MET-inhibitor offered a more profound cell growth inhibition than lapatinib alone [9]. Despite the strong evidence regarding the interplay between MET and HER2 in GC, the current understanding of the regulation of MET expression and activation in relation to lapatinib-resistance in HER2-positive cells requires additional research.
Forkhead box O1 (FOXO1) is a transcription factor and a member of the FOXO subfamily of the Forkhead/winged helix family [10]. Since FOXO1 activates or represses multiple target genes, and consequently regulates a variety of cellular functions [11], dysregulation of FOXO1 would subsequently result in various disease states such as cancer. FOXO1 inactivation has been documented in several cancers, including GC [12], and its association with several anti-cancer drugs has increasing attracted oncologists' attention [13-15].
The existence of a negative crosstalk between FOXO1 and HER2 in parental GC cell lines was previously reported [16]. This crosstalk was associated with cancer cell growth, epithelial-mesenchymal transition, cell migration and invasion in vitro as well as tumorigenicity and metastasis in vivo [16]. However, the relationship between FOXO1 and anti-HER2 drug resistance in GC has not been reported. In the present study, lapatinib-resistant GC cell lines (SNU-216 LR 2-8) were generated by chronic exposure to lapatinib and the potential role of FOXO1 in lapatinib resistance was examined. In addition, we silenced MET expression and investigated its implication in the lapatinib resistance in the lapatinib-resistant, HER2-positive GC cells.
Materials and Methods
1. Cell culture
A HER2-positive GC cell line SNU-216 was purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI 1640 medium (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (FBS; BioWest, Kansas City, MO) in a humidified atmosphere containing 5% CO2 at 37°C.
2. Reagents and antibodies
Lapatinib was purchased from Cell Signaling Technology (Berverly, MA), and cisplatin (CDDP) was purchased from Sigma (St. Louis, MO). Antibodies against phospho-HER2Tyr1221/1222 (pHER2, rabbit monoclonal), HER2 (rabbit monoclonal), phospho-METTyr1234/1235 (pMET, rabbit monoclonal), phospho-AKTSer473 (pAKT, rabbit polyclonal), AKT (rabbit polyclonal), and FOXO1 (rabbit monoclonal) were purchased from Cell Signaling Technology. Antibodies against MET (rabbit polyclonal), β-actin (mouse monoclonal) and secondary antibodies, which are horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
3. Generation of lapatinib-resistant clones SNU-216 LR from SNU-216 cells
SNU-216 cells were cultured in the presence of increasing concentrations of lapatinib over a period of 8 months, reaching a final concentration of 10 μmol/L at the end of this period as described previously [17]. Single-cell clonal populations were obtained from a pool of resistant cells by serial dilutions. Cells were expanded in RPMI-1640 medium containing 10% FBS and lapatinib (1 μmol/L).
4. Growth inhibition assays
The viability of cells was measured indirectly using crystal violet assay as described by Kim et al. [18]. Cells were seeded in 24-well plates at a density of 1×104 cells/well for cell growth and cultured for 4 days. To study the cytotoxicity effect of drugs, 1.5×104 cells were seeded in 24-well plates, incubated for 24 hours, and treated for 3 days at 37°C with indicated concentrations of drugs dissolved in 0.04% dimethyl sulfoxide. Control columns contained cells without drug and blank columns contained medium alone. Medium was aspirated from the wells followed by washing three times with tap water. Attached cells were stained with 0.2% crystal violet aqueous solution in 20% methanol for 10 minutes followed by washing four times and were air dried. Crystal violet stain was dissolved in 10% sodium dodecyl sulfate (SDS) for 10 minutes at room temperature, transferred into 96-well plates, and the absorbance was measured at 570 nm using an enzyme-linked immunosorbent assay reader (Bio-Rad, Hercules, CA). The reading of blank columns was subtracted from each value.
5. Western blot analysis
Western blot analysis was performed as described previously [16]. Proteins were obtained when cells were subconfluent (70%-80%). Briefly, cell lysates in SDS lysis buffer (125 mM Tris-HCl [pH 6.8], 4% SDS, 0.004% bromophenol blue, and 20% glycerol) were separated on 10% SDS–polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride membranes (Millipore Corporation, Billerica, MA) blocked with 5% non-fat dry milk in phosphate-buffered saline–Tween-20 (0.1%, vol/vol) for 1 hour. The membranes were then incubated with a primary antibody against pHER2 (1:1,000), HER2 (1:1,000), pMET (1:1,000), MET (1:1,000), pAKT (1:1,000), AKT (1:1,000), FOXO1 (1:1,000), or β-actin (1:1,000). Horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000) or anti-mouse IgG (1:2,000) was used as a secondary antibody. Enhanced chemiluminescence (Amersham, Arlington Heights, IL) was used to detect the immunoreactive proteins. Equal protein loading was confirmed by β-actin.
6. Transfection of forkhead responsive element–luciferase construct and luciferase reporter assay
To determine FOXO1 nuclear DNA-binding activity in GC cells, luciferase reporter assay was performed as previously described [15]. GC cells were seeded in 24-well plates at a density of 3×104 cells/well and were transiently cotransfected with 0.4 μg forkhead responsive element (FHRE)–luciferase reporter plasmid (reporter construct in which a small region of the Fas ligand promoter contains the three FHREs, Addgene plasmid 1789, Addgene Inc., Cambridge, MA) and 0.4 mg pSV-b-galactosidase vector (Promega, Madison, WI), an internal control, using Lipofectamine Plus (Life Technologies). Twenty-four hours after transfection, assays for luciferase and β-galactosidase were carried out using a Dual-Luciferase Reporter Assay System (Promega). Luciferase activity was measured on an Auto-Lumat LB 9505c luminometer (Berthold Analytical Instruments, Nashua, Germany) and was normalised by β-galactosidase activity.
7. Overexpressing FOXO1 in SNU-216 LR cells
Overexpressing FOXO1 was done by stable transfection of pcDNA3 containing human FOXO1A3 mutant gene (Addgene plasmid 13508, Addgene Inc.). The plasmid FOXO1A3 encodes a constitutively active version of FOXO1 (all three AKT phosphorylation sites are mutated to Ala). Each vector (1 μg) was transfected into GC cells using Lipofectamine Plus according to the manufacturer’s instructions. Twenty-four hours after transfection, G418 (3 μg/mL) was added to select stable FOXO1A3 clones.
8. Lentivirus-mediated short hairpin RNA silencing of HER2
Lentiviral particles containing non-targeting short hairpin RNA (shRNA) or HER2 shRNA were purchased (Sigma). The sequence of HER2 shRNA was 5′-CCGGTGTCAGTATCCA GGCTTTGTACTCGAGTACAAAG CCTGGATACTGACATTTTTG-3′. The control shRNA particles contain 4 bp mismatches within the short hairpin sequence to any known human or mouse gene. Viral infection was performed by incubating GC cells in the culture medium containing lentiviral particles for 12 hours in the presence of 5 μg/mL Polybrene (Santa Cruz Biotechnology). Pooled puromycin (2 μg/mL)-resistant cells were used for further analysis.
9. MET silencing by stable transfection with shRNA plasmid vector
For MET silencing, the pGFP-V-RS plasmid vectors containing either scrambled shRNA or MET shRNA were purchased from OriGene (Rockville, MD). The sequence of MET shRNA was 5′-GCAAGCCAGATTCTGCCGAACCAATGGAT-3′. Each vector (1 μg) was transfected into GC cells using Lipofectamine Plus according to the manufacturer’s instructions. Twenty-four hours after transfection, puromycin (2 μg/mL) was added to select stable MET shRNA clones.
10. Reverse transcription–polymerase chain reaction
Reverse transcription–polymerase chain reaction (RT-PCR) was performed to determine the mRNA level of molecules in GC cells, and the amplification of β-actin transcripts was used as the control to normalize the transcript levels of molecules. Total RNAs were isolated using TRIZOL reagent (Invitrogen, Carlsbad, CA), and reverse-transcription was performed to synthesize cDNAs in a 20 μL reaction mixture containing each gene-specific primer, 1 μg of RNA, 2× reaction buffer, 0.4 μL Taq polymerase, and 1.2 mM MgCl2. The cDNAs of HER2 transcripts were amplified for 28 cycles (30 seconds at 94°C, 30 seconds at 52°C, and 30 seconds at 70°C), the cDNAs of MET transcripts were amplified for 30 cycles (30 seconds at 94°C, 30 seconds at 52°C, and 30 seconds at 72°C), the cDNAs of FOXO1 transcripts were amplified for 25 cycles (30 seconds at 94°C, 1 minute at 57°C, and 1 minute at 72°C), and the cDNAs of β-actin transcripts were amplified for 18 cycles (94°C for 30 seconds, 52°C for 30 seconds, and 70°C for 30 seconds). The PCR cycling numbers had been optimized to avoid the amplification saturation. Five microliter RT-PCR product was separated on 1% agarose gels, which were subsequently stained with ethidium bromide. Primer sequences were 5′-GGGAGAG AGTTCTGAGGATT-3′ and 5′-CGTCCGTAGAAAGGTAGTTG-3′ for HER2, 5′-TTGC CAGAGACATGTATGATAAAG-3′ and 5′-CCAGCATTTTAGCATTACTT-3′ for MET, 5'-GCAGATCTACGAGTGGATGGTC-3’ and 5’-AAACTGTGATCCAGGGCTG TC-3’ for FOXO1, and 5’-ACACCTTCTACAATGAGCTG-3’ and 5’-CATGATGGAGTTGAAGGT AG-3’ for β-actin.
11. Statistical analysis
All experiments were performed using triplicate cultures, and the results were expressed as the mean±standard deviation. Statistical analyses were conducted using GraphPad Prism software for Windows 7 (ver. 4, GraphPad Software, San Diego, CA). Data were evaluated by two-tailed Student’s t test. Differences with a p-value of < 0.05 were considered statistically significant.
Results
1. Lapatinib-resistant, HER2-positive GC cells exhibit downregulation of FOXO1
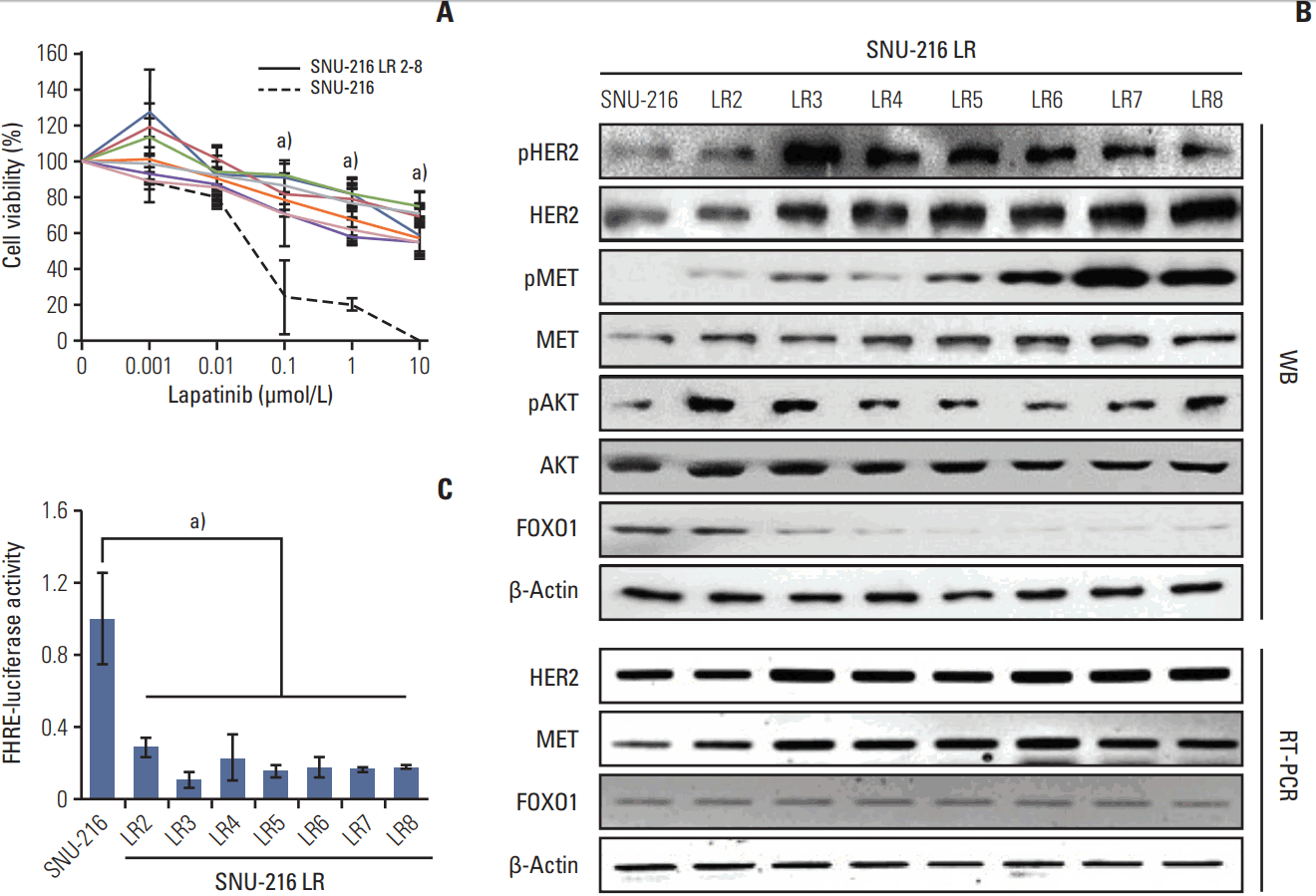
To verify if FOXO1 is involved in the acquired lapatinib resistance in HER2-positive GC cells, stable lapatinib-resistant GC cell lines SNU-216 LR (LR2-LR8) were generated from lapatinib-sensitive parental SNU-216 cells by exposure to continuous treatment with lapatinib (0-10 μmol/L) over a period of 8 months as described previously by Kim et al. [17]. While parental SNU-216 cells treated with 10 μmol/L lapatinib displayed an almost complete abrogation of growth, the resistant cell lines showed significantly lower cell viability reduction than parental cell line (Fig. 1A).
Western blot analysis (Fig. 1B) confirmed HER2 overexpression and low expression of FOXO1 in parental SNU-216 cells as previously reported [16]. In SNU-216 LR cells (except LR2) with the acquired lapatinib resistance, the expression and activation (manifested by phosphorylated forms) of HER2 and MET increased with a more distinctive upregulation of MET. Consistently, phosphorylated AKT (common downstream signaling protein of HER2 and MET), but not total AKT, increased in SNU-216 LR cells. In contrast, FOXO1 expression decreased in most of resistant cell lines (L3-L8). Downregulation of FOXO1 activation in all lapatinib-resistant cell lines was confirmed using the luciferase reporter assay (Fig. 1C). In addition, RT-PCR analysis showed that mRNA expressions of HER2 and MET were increased in SNU-216 LR cells. In contrast, FOXO1 mRNA expression was not changed (Fig. 1B).
2. FOXO1 overexpression reduces resistance to lapatinib
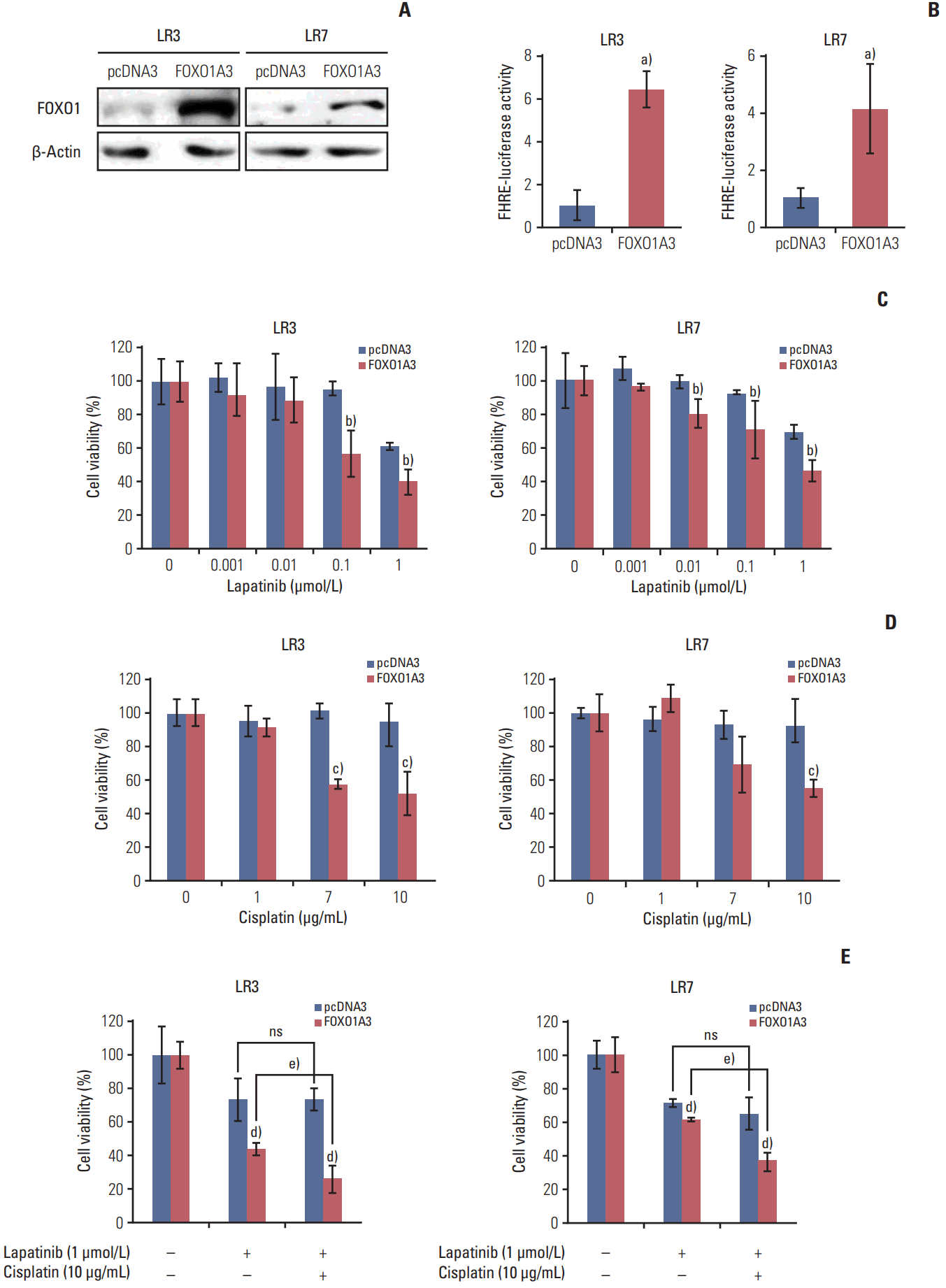
To examine whether FOXO1 is related to the acquired lapatinib resistance in SNU-216 LR cells (LR3 and LR7), FOXO1 expression was modulated by transfection with a construct expressing constitutively active FOXO1 (FOXO1A3). Cells transfected with empty pcDNA3 vector were generated as control. Western blot analysis (Fig. 2A) and the luciferase reporter assay (Fig. 2B) confirmed that FOXO1 expression and transcriptional activity were increased in FOXO1A3-transfected cells compared to vector control cells. The role of FOXO1 in the acquired lapatinib resistance in HER2-positive GC cells was examined by comparing cell growth of SNU-216 LR cells with or without FOXO1 overexpression using crystal violet assay. Treatment of SNU-216 LR3 cells (Fig. 2C) with lapatinib (1 μmol/L) for 72 hours decreased cell viability to ~60% in vector cells and ~40% in FOXO1A3-transfected cells compared to untreated cells. This result demonstrated a significant difference in the lapatinib cytotoxicity between vector control cells and FOXO1A3-transfected cells (p=0.0099). Similar results were shown in SNU-216 LR7 cells (p=0.006) (Fig. 2C).
3. Constitutive FOXO1 activation increases the cytotoxic effect of combined treatment with lapatinib and cisplatin compared to lapatinib alone
In the preliminary study, we found that parental SNU-216 cells were cisplatin-sensitive, but SNU-216 LR cells were cisplatin-resistant (data not shown). Thus, SNU-216 LR cells with lapatinib resistance also developed cross-resistance to cisplatin. However, treatment of SNU-216 LR3 cells with cisplatin (10 μg/mL) in the presence of FOXO1 overexpression significantly decreased cell viability (52% vs. the untreated control) compared with vector control cells (93% vs. the untreated control) (p=0.016) (Fig. 2D). Similar results were shown in SNU-216 LR7 cells (p=0.008) (Fig. 2D).
Although lapatinib plus chemotherapy using parental SNU-216 cells showed an additive or synergistic effect in vitro [19], inconsistent results were shown in the second-line treatment of patients with HER2-positive GC [20]. In the present study, the effect of adding cisplatin to lapatinib in SNU-216 LR3 and LR7 cells in the absence or presence of FOXO1 overexpression was examined. In the presence of FOXO1 overexpression, the combined treatment with cisplatin and lapatinib showed an additive cytotoxic effect (26% vs. the untreated control) in SNU-216 LR3 cells compared to treatment with lapatinib alone (44% vs. the untreated control) (p=0.022). However, there was no difference in cell viability in the absence of FOXO1 overexpression (Fig. 2E). Consistent results were shown in LR7 cells (p=0.001) (Fig. 2E). Thus, FOXO1 induces the efficacy of adding cisplatin to lapatinib in lapatinib-resistant SNU-216 LR cells.
4. FOXO1 transcriptionally downregulates HER2 and MET
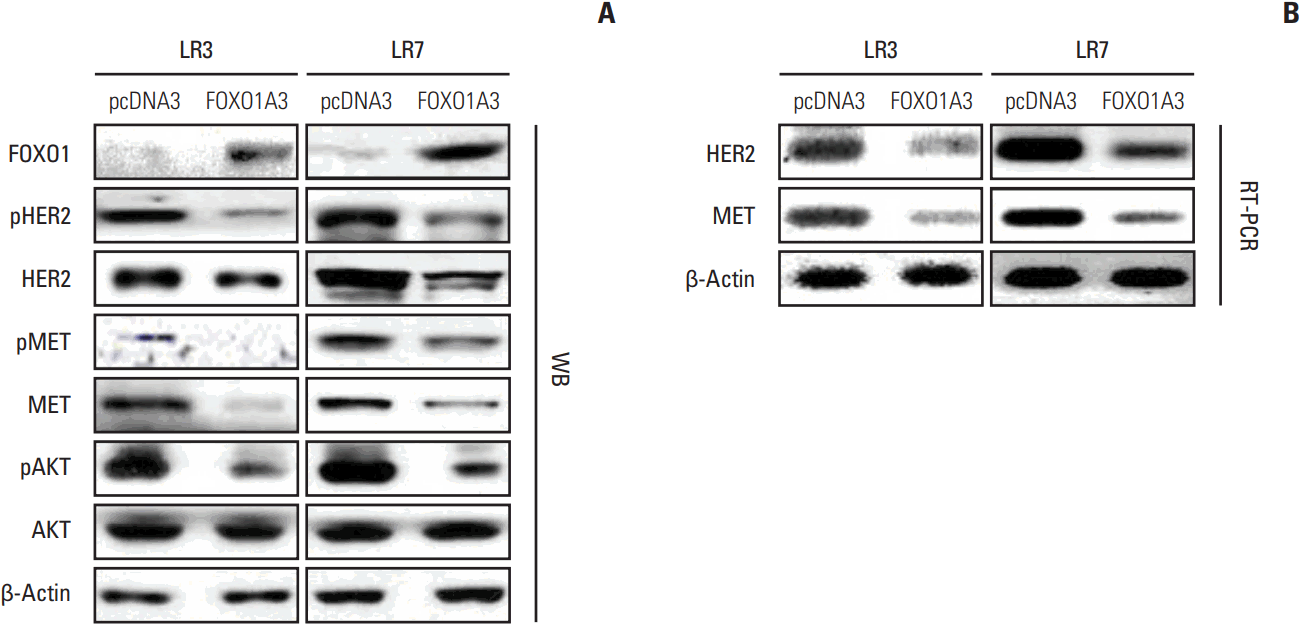
HER2 and MET are strong predictors of lapatinib sensitivity in GC cells [10,11,17]. However, the molecular mechanism underlying a link between HER2 and MET with respect to lapatinib resistance remains undefined. To analyze the relationship between FOXO1 and HER2/MET in SNU-216 LR cells, we increased the FOXO1 expression and activation by transfection of FOXO1A3 as shown in Fig. 2A and B. In addition, Western blot analysis and RT-PCR were performed. Western blot analysis showed that the total and phosphorylated forms of HER2 and MET were downregulated by FOXO1 overexpression (Fig. 3A). Furthermore, the protein expression of phosphorylated AKT, but not the total AKT, was decreased. Consistently, RT-PCR demonstrated that mRNA expressions of HER2 and MET were downregulated (Fig. 3B). Taken together, FOXO1 negatively regulates HER2 and MET expressions at the transcriptional level, suggesting its involvement in the lapatinib-induced HER2/MET signaling pathway.
5. The acquired resistance to lapatinib and/or cisplatin is reversed by silencing HER2
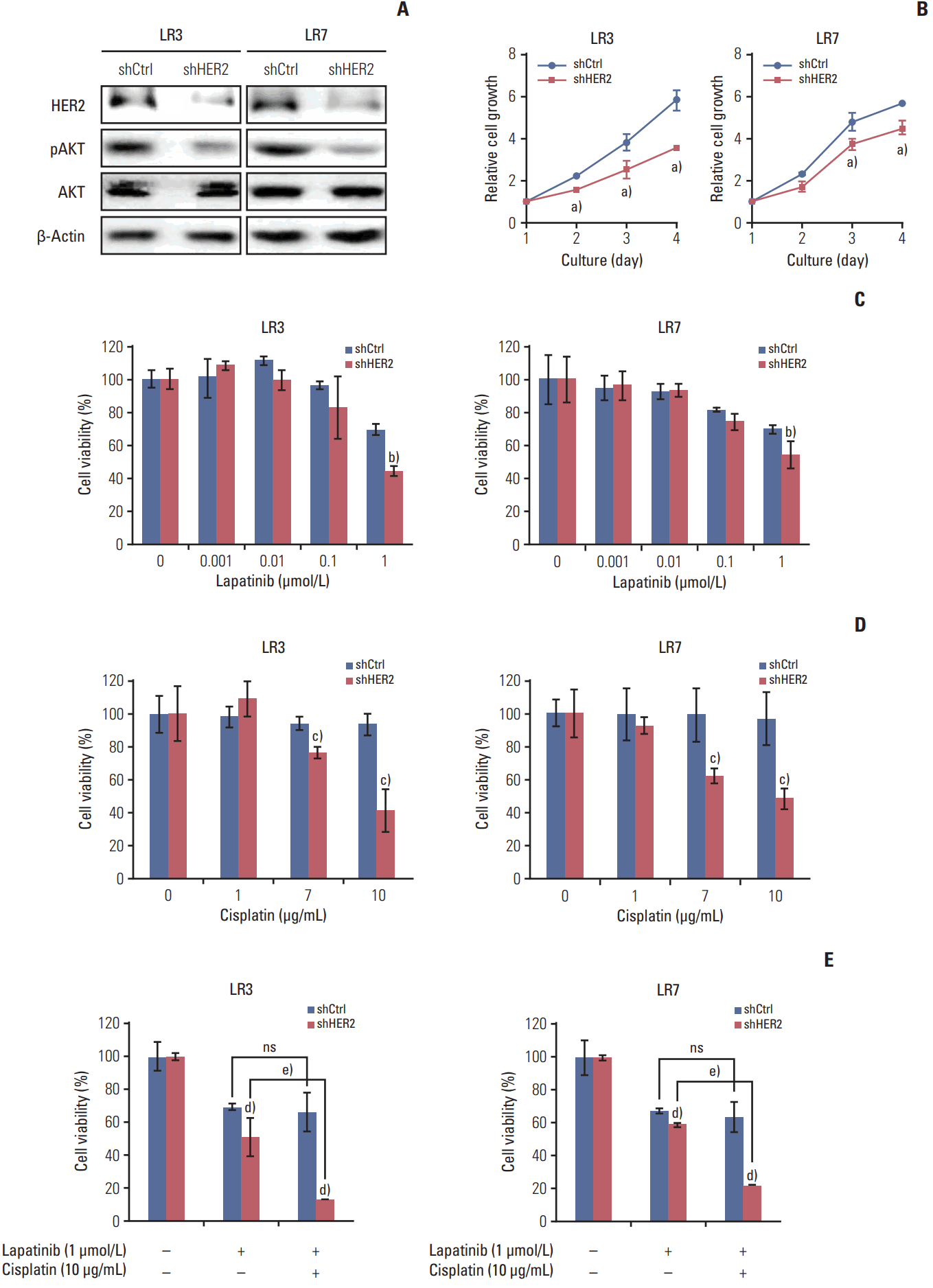
Parental SNU-216 cells are responsive to lapatinib because of the presence of HER2 overexpression. In the present study, however, SNU-216 cells became resistant to lapatinib after chronic exposure to lapatinib in spite of HER2 upregulation (Fig. 1A and B). To investigate whether the dependency of cell viability on HER2 expression persists in SNU-216 LR cells, HER2 was downregulated by RNAi (Fig. 4A). We found that HER2 shRNA-transfected cells had a lower level of pAKT (Fig. 4A) and showed growth inhibition (Fig. 4B). HER2 downregulation also significantly suppressed lapatinib resistance (Fig. 4C) and cisplatin resistance (Fig. 4D) compared to control shRNA cells. Furthermore, the combined treatment of HER2 shRNA-transfected SNU-216 LR cells showed an additive cytotoxic effect compared to treatment with lapatinib alone (Fig. 4E).
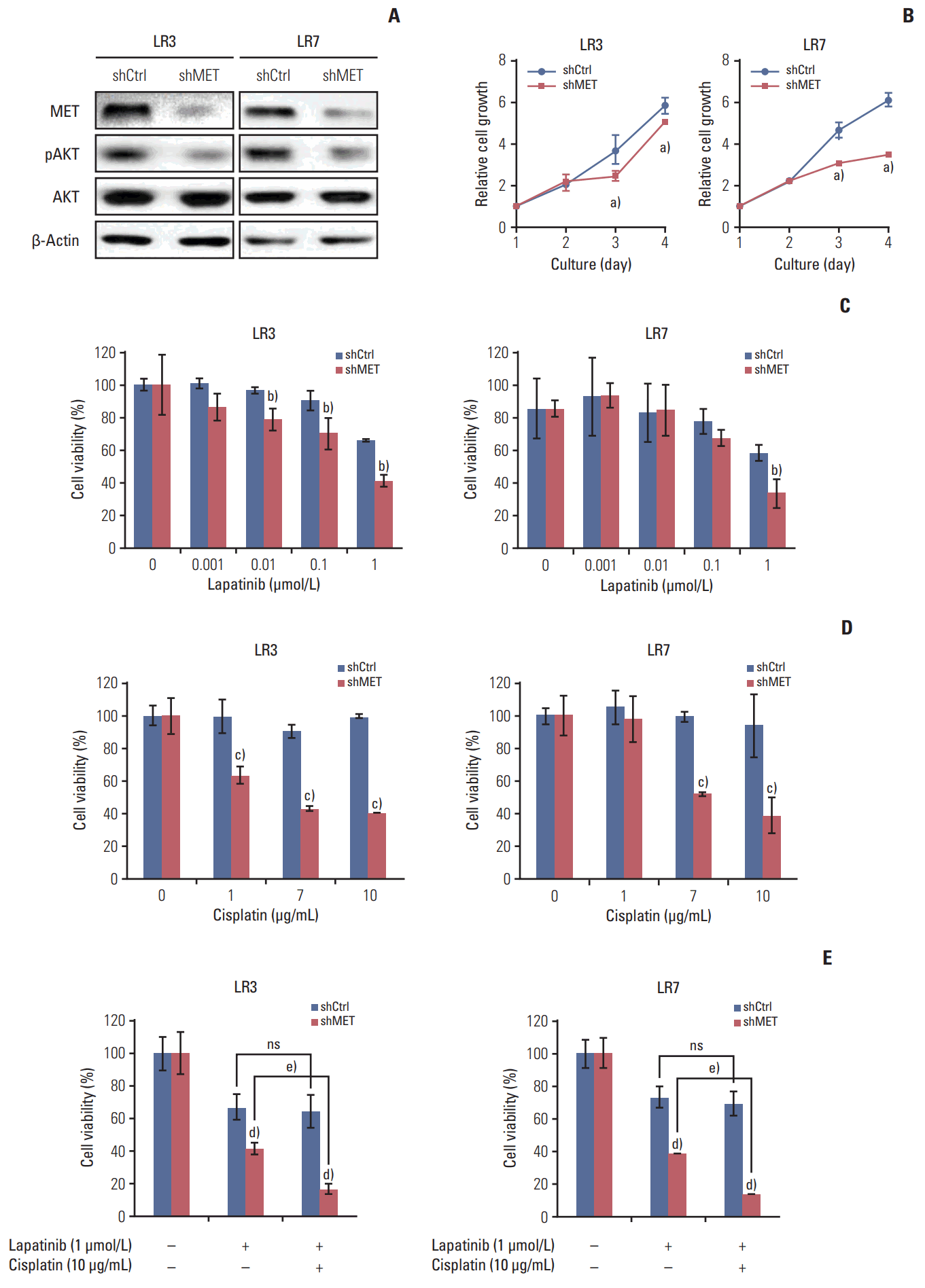
6. The acquired resistance to lapatinib and/or cisplatin is reversed by silencing MET
Parental SNU-216 cells showed a low level of MET expression, which notably increased in SNU-216 LR cells (Fig. 1B). To investigate the role of MET in lapatinib resistance in SNU-216 LR cells, MET expression was downregulated by RNAi (Fig. 5A). MET shRNA-transfected cells had a lower level of pAKT (Fig. 5A) and also showed growth inhibition (Fig. 5B). Furthermore, MET silencing significantly suppressed lapatinib resistance (Fig. 5C) and cisplatin resistance (Fig. 5D) compared to control shRNA cells. In addition, combination treatment showed additional growth inhibition compared to treatment with lapatinib alone in MET shRNA-transfected cells, but not in control shRNA cells (Fig. 5E).
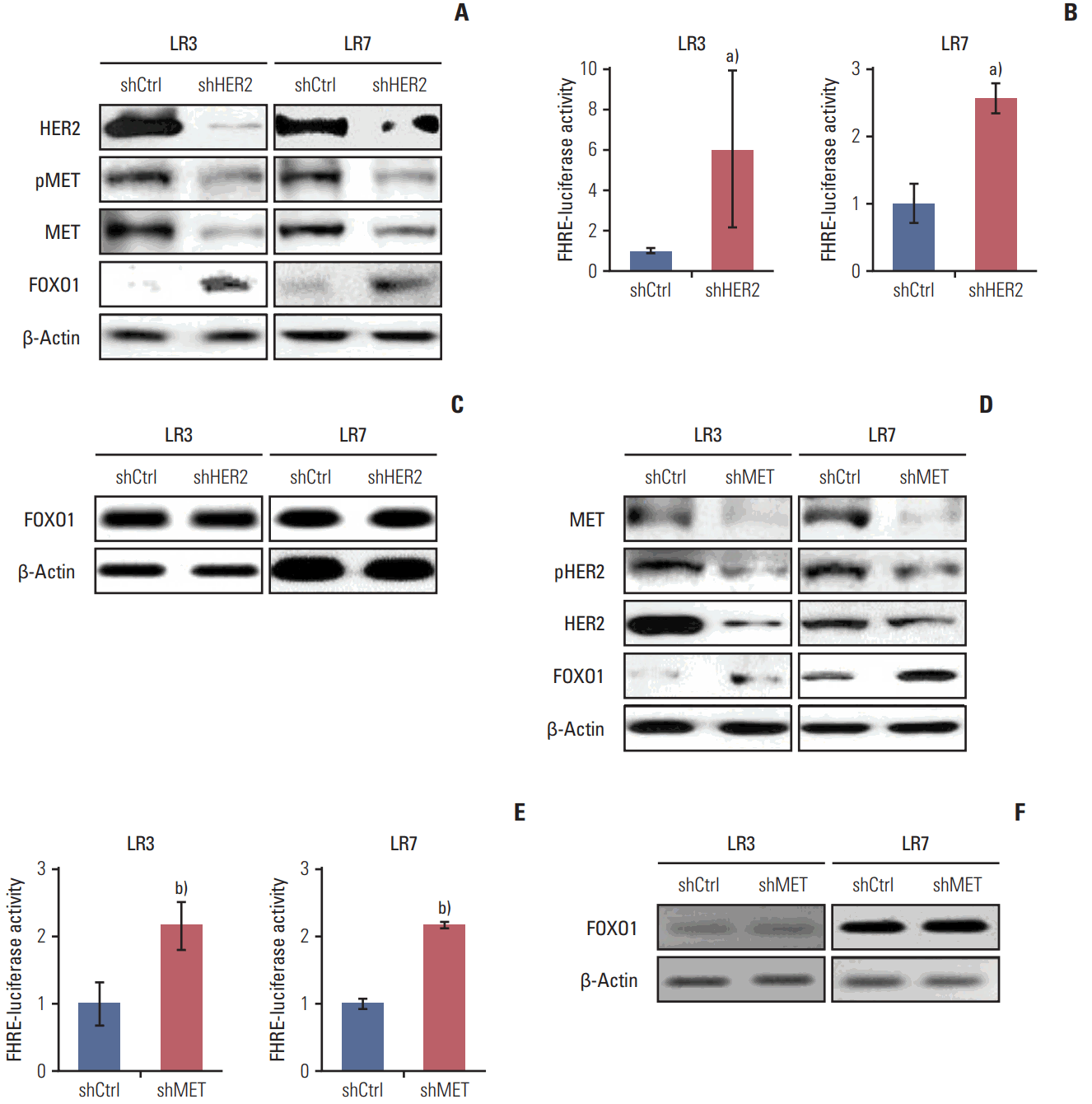
7. HER2 and MET interplay through transcriptional control by FOXO1
To investigate whether interplay between HER2 and MET exists, stable SNU-216 LR3 and LR7 cell lines overexpressing either HER2 shRNA (Fig. 6A) or MET shRNA (Fig. 6D) were used. Western blot analysis showed that HER2 silencing decreased the protein expressions of total and phosphorylated MET (Fig. 6A). In turn, MET silencing reduced the protein expressions of total and phosphorylated HER2 (Fig. 6D). Thus, these findings indicate that there is a positive interplay between these two molecules. Then, the effect of HER2 downregulation on FOXO1 expression and activation was examined. Western blot analysis (Fig. 6A) and luciferase reporter assay (Fig. 6B) demonstrated that HER2 silencing increased protein expression and activation of FOXO1 without a change in FOXO1 mRNA expression as shown by RT-PCR (Fig. 6C). Similar findings were observed in MET shRNA-transfected cells (Fig. 6D-F). These results indicate that FOXO1 expression is negatively regulated at the post-transcriptional level by HER2 and MET.
Discussion
The acquisition of drug resistance in treated patients has become a significant issue in the establishment of strategy for human cancer therapy. Moreover, the complex interplay of signal-transduction pathways further complicates customizing of cancer treatments which might target a single mechanism [21]. The purpose of the present study was to determine the correlation between FOXO1 expression profile and the sensitivity to lapatinib alone or in combination with cisplatin, thereby providing a new strategy for treating lapatinib-resistant, HER2-positive GC. Here, FOXO1 suppression was identified as a determinant of acquired lapatinib resistance in HER2-positive GC cells, at least in part, through negative crosstalks with HER2 and MET. To the best of our knowledge, this is the first study to demonstrate the involvement of FOXO1 in anti-HER2 drug resistance and its association with MET in GC cells.
Given that HER2 serves as a putative target for therapy in HER2-positive GC, elucidating the molecular mechanism of lapatinib resistance is critical to establish a more efficient treatment strategy for patients who failed to respond to adjuvant trastuzumab-based chemotherapy. However, the molecular mechanism underlying the unresponsiveness of GC to lapatinib remains largely unexplained. Previously, lapatinib sensitivity was shown to be positively correlated to the degree of HER2 overexpression in various cancer cells [10]. Consistently, lapatinib responsiveness was shown in HER2-positive parental GC cell lines SNU-216 and NCI-N87 [19]. Although the initially addicting oncoprotein HER2 in parental SNU-216 cells is the target of lapatinib, SNU-216 LR cells were lapatinib-resistant in spite of HER2 upregulation. This suggests that the acquired lapatinib resistance in SNU-216 LR cells may be attributed to an alternative or redundant survival pathway [22]. Indeed, Kim et al. [17] suggested that MET upregulation could confer the acquired lapatinib resistance to lapatinib-sensitive, HER2-positive GC cells.
Previous studies have shown that FOXO1 plays an important role in the regulation of responsiveness of cancer cells to various anticancer drugs [13-15]. For example, FOXO1 increased paclitaxel resistance in ovarian cancer cells [13], adriamycin resistance in breast cancer cells [14] and cisplatin resistance in GC cells [15]. With respect to HER2-positive cancer cells, FOXO1 decreased trastuzumab resistance in HER2-positive breast cancer cells [23,24]. However, different resistance mechanisms have been reported for trastuzumab and lapatinib [25], and lapatinib sensitivity in cultured cells is determined by tissue type [26]. In the present study, FOXO1 activation was downregulated in SNU-216 LR cells compared to parental SNU-216 cells. Thus, the present study hypothesized that FOXO1 is implicated in the acquired lapatinib resistance in these cells in association with HER2 and MET. Taking advantage of lapatinib-resistant, HER2-posistive SNU-216 LR cell lines, we confirmed that lapatinib alone showed weak growth inhibitory effect toward SNU-216 LR cells. Further, FOXO1 overexpression in these cells induced an enhanced cytotoxic effect of lapatinib. These results demonstrate the importance of FOXO1 for the lapatinib-mediated cytotoxic effect in SNU-216 LR cells.
The efficacy of lapatinib alone or in combination with standard chemotherapy for HER2-positive GC is yet to be improved. In the preclinical cell-based study using HER2-positive GC cell lines (SNU-216 and NCI-N87), lapatinib plus chemotherapy showed an additive or synergistic effect [19]. In contrast, a randomized, open-labeled, phase III study (Tytan study) showed that second-line treatment of HER2-positive advanced GC patients with lapatinib plus chemotherapy did not significantly improve overall survival compared to chemotherapy alone [20]. In the present study, the combined treatment with lapatinib and cisplatin did not induce a significant difference in cell viability of SNU-216 LR cells compared to lapatinib treatment, which is consistent with results of the clinical trial. In the presence of FOXO1 overexpression, however, combined treatment resulted in a greater reduction in cell viability compared to treatment with lapatinib alone. These results provides a direct evidence that FOXO1 suppression confers acquired resistance to lapatinib and/or cisplatin in lapatinib-resistant, HER2-positive GC cells. Since FOXO1 involvement in lapatinib resistance has not been previously identified in HER2-positive cancers, this is a novel molecular mechanism underlying acquired lapatinib resistance in HER2-positive GC. These findings provide a basis for the proposal that systemic reintroduction of FOXO1 in HER2-positive GC patients could result in a selective lapatinib toxicity in cancer cells. In the near future, it should be possible to generate an enhanced anti-cancer effect via a combination of lapatinib and FOXO1-replacement therapy. However, the technique of transcription factor-replacement therapy is not yet in general use.
The phenomenon of oncogene addiction has revealed potentially important therapeutic opportunities that can lead to the selective elimination of tumor cells showing dependence on a protein or pathway [22]. Just as acute inactivation of addicting oncoproteins frequently leads to cancer cell death, recent evidence points to similar outcomes induced by the reintroduction of a wild-type version of tumor suppressor genes that are frequently inactivated in cancer cells [22]. Accumulating data on the “addiction to lack of tumor suppressor genes” indicate that, in the establishment of the oncogene addicted state, a prerequisite may involve the removal of support systems such as tumor suppressors (p53, FHIT, PTEN, LKB1, and TESTIN) [22]. In the present study using SNU-216 LR cells, upregulations of oncogenes HER2 and MET as well as downregulation of tumor suppressor FOXO1 were observed. In addition, FOXO1 overexpression reduced mRNA expressions of HER2 and MET. Accordingly, it seems that chronic exposure to lapatinib induced FOXO1 downregulation, and consequently upregulated of HER2 and MET through transcriptional control. Taken together, we speculate that FOXO1 inactivation may be a prerequisite in the establishment of the addiction to HER2 as well as MET in SNU-216 LR cells.
Although lapatinib responsiveness is associated with HER2 overexpression in parental GC cell lines [19], HER2 was upregulated in lapatinib-resistant, HER2-positive GC cell lines in the present study. However, the effect of HER2 modulation on the acquired lapatinib resistance in these cells has never been evaluated. This study investigated whether the loss of HER2 addiction or addiction switching to an alternative oncogene [22] is induced by chronic exposure to lapatinib. HER2 expression in SNU-216 LR cells was downregulated by using RNA interference, which resulted in AKT inactivation and cell growth suppression. These results indicate that HER2 addiction still remains. Inconsistently, HER2 downregulation significantly suppressed resistance to an anti-HER2 drug lapatinib, In addition, cisplatin resistance was decreased in HER2-silenced SNU-216 LR cells, which is consistent with a previous report [27].
MET has been shown to cross-react with EGFR proteins and possibly substitutes for their activity, thus conferring resistance to EGFR-targeting drugs [28]. With respect to GC cells, HGF-induced MET activation in HER2-positive GC cell lines (SNU-216 and NCI-N87) induced lapatinib resistance [10]. In the present and previous [17] studies, chronic exposure to lapatinib upregulated MET compared to parental HER2-positive GC cells. However, the effect of MET modulation on the acquired lapatinib resistance in these cells has not been shown. This study examined whether cancer cells exhibit MET addiction in addition to original HER2 addiction. MET downregulation in SNU-216 LR cells decreased AKT activation and cell growth, which was similar to results obtained with HER2 downregulation. Consistently, MET downregulation significantly suppressed lapatinib and/or cisplatin resistance. Since both HER2 and MET contributed to lapatinib resistance in SNU-216 LR cells, it seems that MET provides redundant survival signals through the activation of downstream survival pathways that overlap with those of HER2. Thus, SNU-216 LR cells can be considered to be “co-addicted” to HER2 and MET.
The present study showed that MET expression and activation were clearly lower than those of HER2 in parental SNU-216 cells. After acquisition of lapatinib resistance, MET was notably upregulated in the majority of SNU-216 LR cell lines compared to HER2. Previously, MET amplification was shown to be responsible for the MET overexpression induced by EGFR RTK inhibitor treatment of non-small cell lung cancer patients who displayed acquired resistance [22]. However, Kim et al. [17] reported that there is no MET gene amplification in HER2-positive SNU-216 LR cells with acquired lapatinib resistance. Although concomitant overexpression of HER2 and MET was observed in a subset of GC patients [9], the regulatory relationship between these two molecules has not been demonstrated in GC. Our results showed that HER2 downregulation in SNU-216 LR cells suppressed the expressions of both total and phosphorylated MET and vice versa. Thus, it seems that the interplay between these two molecules in the survival signaling pathway is not due to transphosphorylation, instead is due to the activations of overlapping downstream molecules. Since FOXO1 is negatively controlled by and controls HER2 and MET, FOXO1 seems to serve as an important linker between HER2 and MET signaling pathways via negative crosstalks. In addition, downregulation of either HER2 or MET increased FOXO1 protein expression and activation, but not mRNA expression, which indicates negative regulation at the post-transcriptional level.
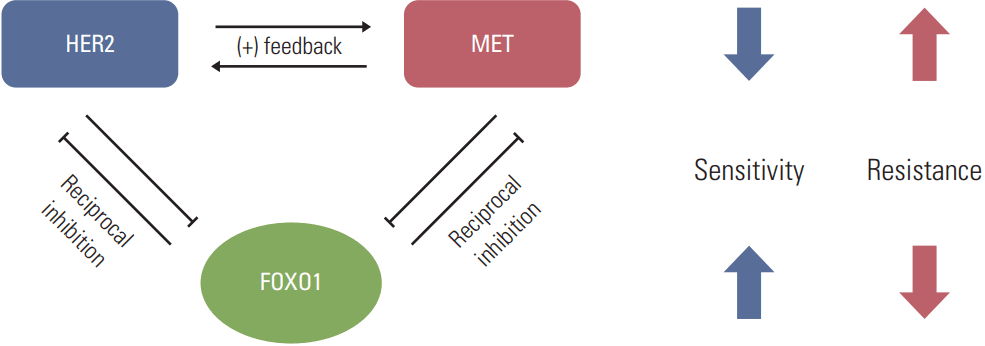
In conclusion, the present study shows a novel molecular mechanism that could cause the acquired lapatinib resistance in a subset of HER2-positive GC cells. It seems that FOXO1 suppression is implicated in the acquisition of lapatinib resistance in HER2-positive GC cells through upregulation of MET as well as HER2 (Fig. 7). Thus, this should be taken into consideration when designing combination therapies for a subset of lapatinib-resistant, HER2-positive GC.
 XML Download
XML Download