

 PDF
PDF Citation
Citation Print
Print


Introduction
Lung cancer is the second most common cancer worldwide, and about 14% of all new cancer patients have lung cancer. In 2010, about 222,500 new cases were reported, and about 157,300 people died from lung cancer in the United States [1,2]. The high mortality of lung cancer may be attributed to the fact that 70% of lung cancer patients are at an advanced stage (stage IV) of the disease at initial diagnosis, and therefore incurable [3]. About 85% of lung cancer cases are non-small cell lung cancers (NSCLC), which are classified into adenocarcinomas (ADC), squamous cell carcinomas (SCC), and large cell carcinomas based on the major histological subtype [2]. Until recently, the various subtypes of NSCLC were grouped together for the purpose of treatment, but it is now widely known that different histologic subtypes should be treated as separate disease entities [2,4]. SCC is the second most prevalent type of lung cancer. However, because the molecular pathogenesis of this disease is not well understood, no approved targeted therapeutics are available for its treatment [5]. Therefore, performing comparative analyses of lung SCCs and identifying potential therapeutic targets would lead to significant growth in cancer treatments. As a result, a great amount of progress has been made in studies and clinical trials to develop targeted treatments for lung cancer patients. Mutations in the epidermal growth factor receptor (EGFR) and KRAS are the most common causes of lung cancer [6], and although the frequency of echinoderm microtubule-associated protein-like 4–anaplastic lymphoma kinase (EML4-ALK) rearrangements in NSCLC is low, ALK rearrangements have been reported to be successful targets of specific tyrosine kinase inhibitors such as crizotinib and ceritinib [7]. Mutations of human epidermal growth factor receptor 2 (HER2) and tyrosine-protein kinase MET, have also been found in NSCLCs, but they are rare and their significance is unclear [8,9]. A recent study reported amplified fibroblast growth factor receptor 1 (FGFR1) in 10%-20% of SCCs, suggesting that targeting FGFR1might be a promising therapeutic strategy [10]. However, FGFR inhibitors are not currently in clinical use for the treatment of lung cancers [11]. The use of histone deacetylase inhibitors for SCC treatment has been suggested, as they induced tumor cell death by up-regulating the pro-apoptotic B-cell lymphoma-2 (Bcl-2) family members [12]. In 2011, Hammerman et al. [3] identified novel somatic mutations in the discoidin domain-containing receptor tyrosine kinase 2 (DDR2) gene at a frequency of 3.8% (n=11) in a set of 290 lung SCC samples. DDR2 is a collagen binding receptor, and a subset of DDR2 mutants in cancer are oncogenic and have been shown to promote cell migration, proliferation, and survival [3,13,14]. DDR2 mutations are mainly found in SCCs of NSCLC and are distributed throughout the gene, including in L63V, I120M, and D125Y in the collagen-binding discoidin 1 domain, C580Y, I638F, T765P, G774E, and G744V in the kinase domain, L239R and G253C in the discoidin 2 domain, and G505S in the cytosolic juxtamembrane domain [3,15]. DDR2 L63V and I638F mutations have been shown to confer transforming abilities to NIH3T3 cells, and lung cancer cells with DDR2 L239R or I638F mutations were also shown to be sensitive to treatment with a DDR2 inhibitor, dasatinib [3]. Therefore, identifying mutations in DDR2 could provide candidates to modulate the response of lung SCCs to chemotherapy [16]. In this study, we analyzed 100 lung SCC samples from Korean patients to identify novel somatic mutations in DDR2 and investigated the frequencies and functions of these mutations.
Materials and Methods
1. Case selection
The study sample was composed of 100 patients with SCC of the lung. Ninety-seven patients underwent biopsy and/or surgical resection at Samsung Medical Center (Seoul, Korea) between 2000 and 2011. DDR2 mutation analyses were performed mainly on the biopsy specimens, which were from a total of 85 cases. Twelve cases were obtained by surgical resection and three cases were received from an outside hospital. Clinical data including age, sex, and stage were obtained from patient records (Table 1). Slides were reviewed independently by two pathologists to verify the diagnoses and tumor content in the tissues. This study was approved by the Institutional Review Board of Samsung Medical Center.
Table 1.
Clinicopathologic parameters of squamous cell lung cancer patients
![]()
2. DDR2 DNA sequence analysis
Genomic DNA was extracted from paraffin-embedded tissues using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany). We subjected 100 ng of genomic DNA to polymerase chain reaction (PCR) amplification of exons 6, 15, 16 and 18 of the DDR2 gene. The primer pairs used to amplify the complete coding sequences of DDR2 exons 6, 15, 16, and 18 are outlined in Table 2. PCR was performed on 20 μL reaction mixtures containing 100 ng of template DNA, 2 μL 10× PCR buffer, 0.25 mM dNTP, 10 pmol primers, and 1.25 U Taq DNA polymerase (iNtRON, Seongnam, Korea). PCR products were separated via electrophoresis on 2% agarose gels and purified using the QIAquick PCR purification kit (Qiagen). Bidirectional sequencing was performed using the BigDye Terminator v1.1 kit (Applied Biosystems, Foster City, CA) on the ABI 3130XL genetic analyzer (Applied Biosystems). Sequencher ver. 4.10.1 (Gene Codes Corporation, Ann Arbor, MI) was used along with a manual review of chromatograms for sequence analysis. Confirmatory re-sequencing from replicate PCR amplification reactions was performed for any sequence that was ambiguous or that deviated from the wild type, so that all abnormal sequences were verified in at least quadruplicate for replicate amplification reactions.
Table 2.
Primer sequences and polymerase chain reaction conditions
![]()
3. Cell culture
NIH 3T3 cells were obtained from the Korean Cell Line Bank and BEAS-2B (human bronchial epithelium, ATCC CRL-9609) cells were obtained from the American Type Culture Collection (Manassas, VA), Both two cell lines were expanded in Dulbecco’s modified Eagle’s medium (JBI, Daegu, Korea) supplemented with 10% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA), 100 units/mL penicillin, and 100 mg/mL streptomycin (Life Technologies). To generate NIH 3T3 and BEAS-2B stable cells expressing DDR2 wild-type (WT) and mutants, cells were transfected with LacZ (negative control), DDR2 WT, L63V, V582E, L595P, or I638F DDR2 mutant expressing lentiviruses for 24 hours and then selected with 6 μg/mL blasticidin for 2 weeks.
4. Cloning DDR2 constructs
For the full-length DDR2 constructs, we used cDNA synthesized from Hs578T cell line as a template and DDR2 WT and mutant constructs were generated using PCR reactions. We described primer sets for cloning of DDR2 constructs in the Table 2. The amplified DDR2 wildtype PCR product was cloned into gateway entry vector pCR8/GW/TOPO (Invitrogen, Thermo Fisher Scientific Inc., Carlsbad, CA). DDR2 mutants were generated using the EZchange Site-directed Mutagenesis kit (Enzynomics, Daejeon, Korea) with the following mutagenic primer sets; all mutations were verified by Sanger sequencing. The WT and mutant DDR2 constructs were transferred into pLenti6.3/V5-DEST gateway destination vectors (Invitrogen, Thermo Fisher Scientific Inc.) following the manufacturer’s instructions. The pLenti-based expression vector and the ViraPower Packaging Mix vector (Invitrogen, Life Technologies) were co-transfected into the 293FT cell line to produce a lentiviral stock following Life Technologies protocols.
5. Western blotting
Whole cell lysates from cells expressing LacZ, WT, L63V, V582E, L595P, or I638F were prepared using a modified RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, and 0.25% sodium deoxycholate) and a protease inhibitor cocktail (GenDepot, Barker, TX). The lysates were centrifuged at 15,000 ×g for 30 minutes at 4°C and diluted with 4× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDSPAGE) sample buffer (40% glycerol, 240 mM Tris/HCl pH 6.8, 8% sodium dodecyl sulfate, 0.04% bromophenol blue, 5% beta-mercaptoethanol). All samples were boiled at 95°C-100°C for 10 minutes, and loaded in the SDS-PAGE on an 8%-10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Merck Millipore, Billerica, MA). Blots were blocked with 5% w/v nonfat dry milk (BD, Transduction Laboratories, San Diego, CA) for 1 hour before primary antibodies were added to the blocking solution and incubated overnight at 4ºC on a shaker. The blots were probed with an anti-DDR2 antibody (2538-DR, R&D Systems Inc., Minneapolis, MN), and anti–α-tubulin, –β-actin antibodies (sc-8035, Santa Cruz Biotechnology), and anti–phosphotyrosine-100, –Src and c-Src antibodies (Cell Signaling Technologies, Danvers, MA). Blots were washed 2-3 times with TBST buffer (Tris-buffered saline and Tween 20) and then incubated with the horseradish peroxidase (HRP)–conjugated anti-goat or rabbit IgG secondary antibodies for 1 hour followed washed with TBST for 1 hour. Antibody detection was performed using chemiluminescent HRP substrate (EMD Millipore, Billerica, MA). All western blot images are representative of at least three independent experiments.
6. Proliferation assay
Cell proliferation rates were measured using an EZ-Cytox cell viability assay kit (Daeil Lab Service, Seoul, Korea) according to the manufacturer’s instruction. BEAS-2B cells expressing the target gene were seeded into 96-well plates at a density of 3×103 cells per well. After 72 hours, 10 mL of EZ-Cytox reagent was added to each well and incubated for 2 hours at 37ºC. The absorbance was measured at a wavelength 450 nm (foreground) and 650 nm (background) using a spectrophotometer.
7. In vitro transforming assay
Analysis of the transforming activity of kinase fusions was performed with Matrigel (BD Biosciences, Bedford, MA) and soft agar. BEAS-2B stable cells expressing DDR2 WT, L63V, V582E, L595P, or I638F mutant were cultured in Matrigel. The bottom layer of each well was coated with 30 μL of Matrigel and allowed to gel by incubating for 30 minutes at 37ºC. Then, 10,000 cells resuspended in 150 μL of Matrigel were loaded onto the bottom layer of each well. Medium with 10% FBS was then overlaid onto the gel and replaced every other day. The images of transformed foci were obtained after culturing for 7 days. For the soft agar assay, the base layer of each well consisted of 1.5 mL of medium with a final concentration of 0.5% Noble agar (BD Biosciences). After bottom agar solidification, 1.5 mL of 0.35% agar containing NIH3T3 cells (20,000) was seeded on the bottom agar layer and incubated for 14 days. Medium was changed every 2 days for 2 weeks. Colonies were fixed with 4% paraformaldehyde and then stained with 0.05% crystal violet (Sigma-Aldrich, St. Louis, MO), and representative images were taken by a phase-contrast microscope (Olympus CKX41, Tokyo, Japan) using i-Solution Lite image analysis software (Image & Microscope Technology, Daejeon, Korea). Statistical significance was analyzed by the GraphPad Prism 5 (ver. 5.01, GraphPad Software, Inc., La Jolla, CA).
Results
Among 100 patients, two patients were found to have DDR2 mutations (c.1754T>A [p.V582E] and c.1784T>C [p.L595P] on exon 15). PolyPhen analysis (http://genetics.bwh.harvard.edu/pph/) predicted that these variants would damage the structure and function of DDR2 (Table 3).
Table 3.
Predicted impacts of amino acid substitutions on the function of variants of DDR2
![]()
A 71-year-old male patient (case 1) underwent a follow-up chest computed tomography (CT) scan in March 2009 for chronic obstructive pulmonary disease that was diagnosed in 1998. The patient was a smoker with a 92-pack/yr history. Chest CT showed a 3 cm nonenhancing mass-like consolidation in the central portion of the left lower lobe with a background of patchy fibrotic changes, as well as scattered calcified and noncalcified sub-centimeter nodules in both lungs (Fig. 1A). A bronchoscopic biopsy performed in May of 2009 revealed high-grade squamous dysplasia. A chest CT in September of 2010 showed that the mass-like consolidation had increased in size to 6.2 cm and that there were numerous new cavitary and noncavitary nodules in both lungs (Fig. 1B). These nodules were suspected to be primary lung cancers with intra-lung metastases. Hilar, mediastinal, and supraclavicular lymph nodes were suspected to have metastases by capsular invasion. A second bronchoscopic biopsy revealed tumor cells of squamous differentiation with intercellular bridges (Fig. 1C). No mutations were observed in EGFR or KRAS. The patient refused palliative chemotherapy and only received supportive care. The patient was lost to follow-up after January 2011. When DDR2 mutation analysis was performed for this patient, c.1745T>A (p.V582E) on exon 15 was identified (Fig. 1D).
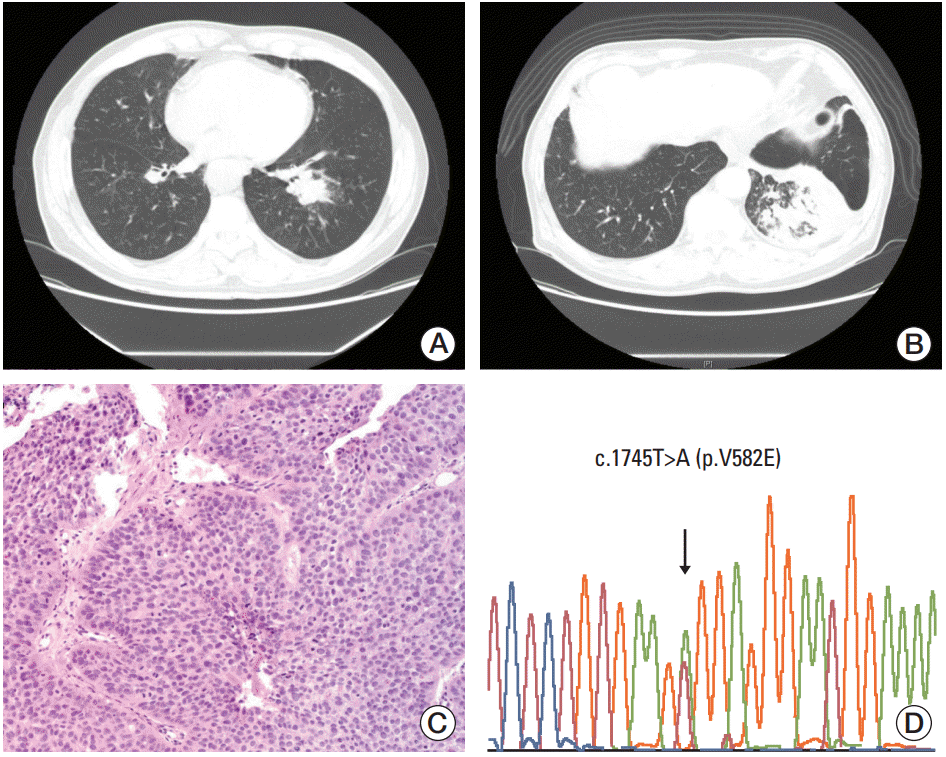
Fig. 1.
Radiological and histological features and discoidin domain-containing receptor tyrosine kinase 2 (DDR2) mutation analysis of case 1. (A) Chest computed tomography (CT) in 2009 shows a 3 cm nonenhancing mass-like consolidation with a background of patchy fibrotic changes and scattered calcified and noncalcified sub-centimeter nodules in both lungs. (B) Chest CT in 2010 shows that the mass-like consolidation has increased in size to 6.2 cm. Numerous new cavitary and noncavitary nodules can be seen in both lungs. (C) Squamous cell carcinoma on hematoxylin and eosin section (×200). (D) Chromatogram of DDR2 shows a missense mutation, as depicted by the arrows, with a change in the amino acids from GTG (valine) to GAG (glutamic acid) on exon 15.
![]()
In another case, a 70-year-old male patient (case 2) visited an outside hospital in May of 2010 due to a persistent dry cough. The patient was a 50-pack/yr smoker who had recently ceased smoking. The patient had undergone regular examinations for rheumatoid arthritis over the last 20 years at an outside hospital, and had been under medication, including steroids, for several years. He was diagnosed with diabetes mellitus 1 month prior to his admission at our hospital. A chest radiograph revealed abnormal findings and the patient was referred for further evaluation. At the Samsung Medical Center, chest CT and magnetic resonance imaging were performed and a 5.3 cm necrotic cavitary mass was revealed in the basal segment of the right lower lung lobe without enlarged lymph nodes (Fig. 2A). The mass was diagnosed as SCC by bronchoscopic biopsy (Fig. 2B and C). The patient underwent a lobectomy of the right lower lobe and an en bloc wedge resection of the right middle lobe because the tumor was located in the right lower lobe beside the middle lobe. Following surgery, pathologic examination revealed a 5 cm mass without pleural invasion. We found lymphatic and perineural invasion, but no metastasis, in the 11 lymph nodes examined. The tumor showed keratinized squamous differentiation with intercellular bridges. Upon immunohistochemical staining, we found that the tumor cells were positive for p63 and negative for thyroid transcription factor-1 (Fig. 2D and E). No mutations were observed in EGFR or KRAS. The patient was treated with adjuvant chemotherapy consisting of vinorelbine and cisplatin. He visited an emergency room because of severe back pain 7 days after the initiation of chemotherapy and was subsequently lost to follow-up. DDR2 mutation analysis of this patient revealed c.1784T>C (p.L595P) on exon 15 (Fig. 2F).
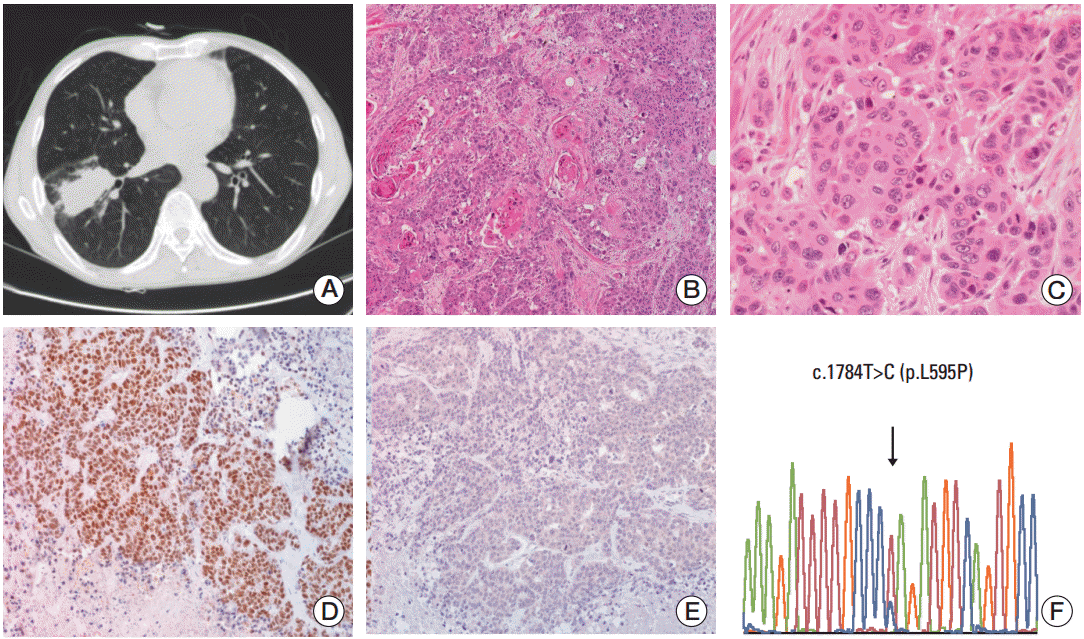
Fig. 2.
Radiological and histological features and discoidin domain-containing receptor tyrosine kinase 2 (DDR2) mutation analysis of case 2. (A) Chest computed tomography scan showing a 5.3-cm necrotic cavitary mass. (B, C) Squamous cell carcinoma on hematoxylin and eosin section (×40 and ×200, respectively). (D) p63 staining showing nuclear staining of tumor cells. (E) Thyroid transcription factor 1 staining showing negative staining of tumor cells. (F) Chromatogram of DDR2 showing a missense mutation, as depicted by the arrows, with a change in the amino acids from CTA (leucine) to CCA (proline) on exon 15.
![]()
To determine if the novel DDR2 mutations identified in this study had oncogenic functions, we constructed lentiviral expression vectors containing the V582E or L595P mutations of DDR2, as well as L63V or I638F mutations as positive controls, as described in previous studies [3]. We then generated clonal populations of NIH3T3 cells stably expressing these constructs. The ectopic DDR2 mutant expression was evaluated using western blot analysis (Fig. 3B). We also showed that cells expressing the mutant DDR2 had increased phosphorylation of the DDR2 receptor (black arrow in Fig. 3B) and Src, relative to control LacZ after collagen stimulation (Fig. 3B). To further validate the tumorigenic potential of DDR2 mutants, we tested their tumor-forming ability in vitro. Interestingly, both cells that expressed DDR2 V582E mutant and those that expressed L595P mutants formed significantly larger colonies, as well as a higher number of colonies than cells harboring DDR2 I638F or L63V mutations (Fig. 3C). Considering the genetic background of patients with the DDR2 mutations, we generated BEAS-2B cells expressing DDR2 wild type and mutants from normal human bronchial epithelium and conducted a cell proliferation assay (S1 Fig. A). As shown in S1 Fig. A, cells expressing DDR2 V582E or L595P mutants showed higher growth rates than control cells. These results indicate that the DDR2 V582E or L595P mutation induced cell proliferation and enhanced tumor progression.
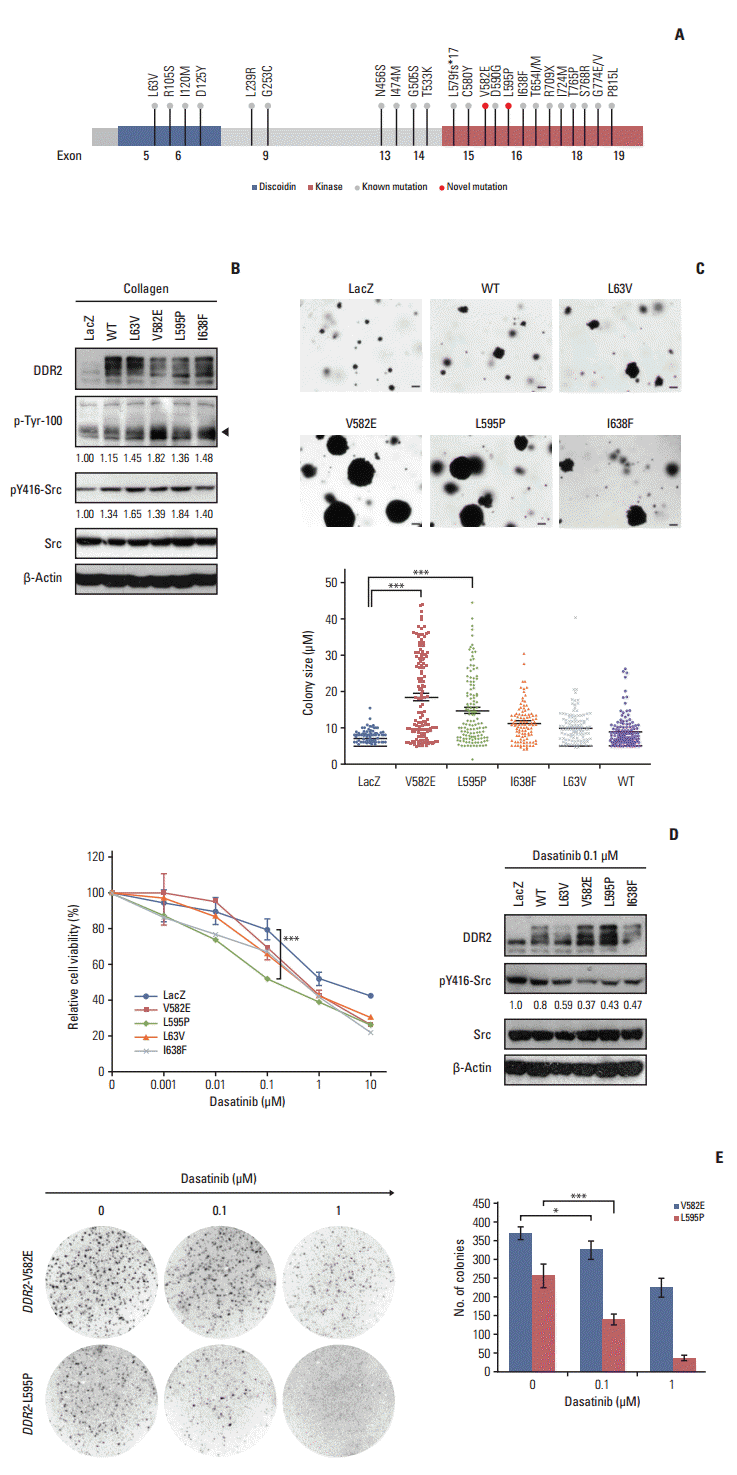
Fig. 3.
Functional effects of discoidin domain-containing receptor tyrosine kinase 2 (DDR2) mutants observed through in vitro assays. (A) Schematic view shows a known mutation and novel mutations (red circles) within the kinase domain of DDR2. (B) Activation of DDR2 and Src molecules by the ectopic expression of DDR2 point mutants. NIH3T3 cells were stably expressed with the indicated DDR2 mutant constructs, stimulated with 10 μg/mL collagen, and then analyzed using western blot. WT, wild type. (C) In vitro transforming assay in soft agar. NIH3T3 cells expressing the indicated mutant protein were seeded in 6-well plates and cultured for 14 days in soft agar. The number of colonies formed per well, indicated as the mean±standard deviation, from the three wells in one representative experiment out of three. ***p < 0.001, scale bars=10 μm. (D) The effect of dasatinib in cells expressing DDR2 mutants. Dasatinib caused reduced proliferation and a decrease in phosphorylation of Src in cells expressing DDR2 V582E and L595P. In the left panel, the NIH3T3 cells expressing the DDR2 mutants were treated with the indicated doses of dasatinib for 72 hours, and cell viability was determined. ***p < 0.001. In the right panel, cells were treated with 0.1 μM dasatinib for 24 hours, followed by cell lysis and detection of the indicated protein using western blot. (E) In vitro colony-forming ability of cells expressing DDR2 mutants following dasatinib treatment. NIH3T3 cells expressing the DDR2 mutants were seeded in soft agar and cultured with or without dasatinib for 7 days. The colonies formed were stained with 0.001% crystal violet and the number of colonies was estimated. The images were obtained using a phase-contrast microscope at 40× magnification. The values shown represent the average of three independent experiments and error bars indicate standard deviations. *p < 0.05, ***p < 0.001.
![]()
Dasatinib is a multiple kinase inhibitor that has been shown to be effective at reducing the tumor size of SCCs expressing DDR2 I683F or L239R mutants [3]. To investigate whether the novel DDR2 mutants were potential therapeutic targets of dasatinib, the relative cell viability was assessed for 72 hours after treatment with 0.01, 0.1, 1, and 10 μg/mL of dasatinib (Fig. 3D). We found that cells expressing DDR2 L595P mutants were more sensitive to 0.1 μg/mL dasatinib than those with DDR2 V582E or other DDR2 mutants, and that the phosphorylation of Src was down-regulated by dasatinib treatment in NIH3T3 cells (Fig. 3D). We obtained the same results for BEAS-2B cells expressing DDR2 L595P mutants as well (S1 Fig. B). Cells were seeded in 3-D Matrigel and cultured for 7 days with or without 0.1 μM dasatinib. Dasatinib treatment caused a decrease in the colony size of cells expressing DDR2 L595P mutant compared with the LacZ control (S1 Fig. B). These results showed that both DDR2 V582E and L595P mutants were oncogenic mutations and cancer drivers. In addition, the DDR2 L595P mutant was dasatinib-sensitive, indicating it might be a potential therapeutic target.
Discussion
The most targetable mutant kinases, EGFR, ALK, and HER2, are mainly found in lung ADCs and only rarely in other histological subtypes. Patients with SCC, who comprise about 40% of all NSCLC cases, are very rarely responsive to targeting agents, and specific genetic alterations in SCC have not been identified to date. Recent molecular analyses have identified genes that may play important roles in lung squamous cell tumorigenesis, including FGFR1, PIK3CA (phosphoinositide-3-kinase catalytic alpha polypeptide), SOX2, and DDR2 [3,22,23]. Tumor cell lines harboring DDR2 mutations have shown increased sensitivity to multiple tyrosine kinase inhibitors in vitro and in vivo, including small molecule inhibitors such as dasatinib, nilotinib, and AP24534 [3,16].
DDR2 mutations have been reported in multiple tumor types with high incidence rates, including gastric carcinomas, bladder carcinomas, melanomas, colorectal cancers, and head and neck cancers [15,19]. Moreover, DDR2 mutations have been reported with 2%-5% frequency in lung SCCs [3,15,19,24]. However, the observed frequency of DDR2 mutations may be associated with ethnic differences in sample populations [25]. For example, no DDR2 mutations were found in a screen of 166 SCC biopsies from Japanese patients [25], but DDR2 mutations were identified with a frequency of 4.6% in a study with 86 Chinese patients with lung SCC [26]. In the present study, we identified a lower mutation frequency of 2% in 100 Korean lung SCC samples. This is likely because our study had a small patient cohort size, but it is not clear if this was due to population differences or other causes.
DDR2 is a potential therapeutic target for the treatment of SCCs in lungs, but genetic alterations such as overexpression of DDR2 or copy number aberrations of the DDR2 locus (1q12-23) have not been reported [3]. However, DDR2 mutations have been associated with responses to targeted agents or to sh-RNA–mediated depletion of DDR2 [3,20,27]. DDR2 mutations are mainly distributed in both the kinase and discoidin domains to recognize specific sites in the extracellular region that binds to collagen, and to activate downstream molecules including SHP-2 as well as SRC and mitogen-activated protein kinases [18,28]. The activation of DDR2 by mutations such as L63V, I638F, and S768R has been associated with cancer progression, including cell proliferation, migration, transformation, and differentiation [3,18]. However, DDR2mutations have also been reported to act as a tumor suppressors in the presence of its ligand, collagen. Kazuto et al. [29] performed the first functional analysis of the DDR2 E655K mutation and found that DDR2 E655K protein bound to the ubiquitin ligase E3 (Cbl-b), and consequently promoted cancer progression by decreasing the growth-inhibiting effects of collagen in NSCLCs. Therefore, identification of DDR2 mutations and investigation of their functions would help to reveal the underlying causes of lung cancer. In this study, we identified cases of DDR2 c.1745T>A (p.V582E) and c.1784T>C (p.L595P) mutations and evaluated the biological function of these mutations in NIH-3T3 and BEAS-2B cells. Our experimental findings have shown that these mutations have transforming activities, and that the DDR2 L595P mutation is a potential therapeutic target of dasatinib (Fig. 3). However, in a 3D proliferation assay using BEAS-2B cells with DDR2mutations, cells with DDR2 V582E mutation did not respond to dasatinib treatment, unlike DDR2 L595P mutant cells (S1 Fig. B). These findings indicate that the DDR2 V582E mutation site may not be an appropriate therapeutic target of dasatinib but could be a suitable target for other multiple kinase inhibitors. Table 4 shows the reported cancer-associated DDR2 mutation sites and their responses to dasatinib treatment.
Table 4.
Cancer-associated DDR2 mutations and responses to dasatinib
| Domain | Mutation site | Source | Dasatinib response | Reference |
|---|---|---|---|---|
| DS | L63V | Primary SCCs | Sensitive | [3, 15] |
| DS-like | L239R | HCC-366 | Sensitive | [3] |
| G253C | Primary SCCs | Sensitive | [3] | |
| IJXM | I474M | Primary HNSCCs | Sensitive | [17] |
| G505S | Primary SCCs | Sensitive | [18] | |
| KD | V582E | Primary SCCs | Sensitive | Present study |
| D590G | Primary HNSCCs | Sensitive | [17] | |
| L595P | Primary SCCs | Sensitive | Present study | |
| I638F | NCI-H2286 | Sensitive | [3] | |
| T654I | HCC-366a) | Resistance | [19, 20] | |
| T654M | HCC-366a) | Resistance | [19, 20] | |
| R709X | Primary HNSCCs | Sensitive | [17] | |
| I724M | Primary HNSCCs | Sensitive | [17, 18] | |
| S768R | Primary SCCs | Sensitive | [3, 21] | |
| G774E | Primary SCCs | Sensitive | [3] | |
| G774V | Primary SCCs | Sensitive | [3] |
![]()
In this study, we investigated the overall frequency of DDR2 mutations in a large clinical sample. We found two novel mutations, V582E and L595P, which play functional roles in cancer progression. Identifying DDR2 mutations and studying their effects would facilitate patient screening during clinical trials of lung SCCs. Our study provided in vitro evidence that DDR2 mutations could be potential therapeutic targets for the corresponding receptors of various tyrosine kinase inhibitors [3,20].
 XML Download
XML Download