

 PDF
PDF Citation
Citation Print
Print


Introduction
The ETV6/RUNX1 rearrangement, t(12;21)(p13;q22), is the most common recurrent genetic abnormality in childhood acute lymphoblastic leukemia (ALL) [1]. Patients with this abnormality are known to have favorable outcome, with event-free survival (EFS) ranging from 80% to 97% reported in recent studies [2-5].
Although children with ETV6/RUNX1 rearrangement may be broadly categorized as a subgroup of precursor B ALL with good prognosis, the subgroup itself could be heterogeneous in terms of additional chromosomal and genetic abnormalities found concurrent with ETV6/RUNX1. Additional chromosomal abnormalities found in patients with ETV6/RUNX1 include loss of the second ETV6 allele, gain of RUNX1, duplication of the derivative chromosome 21, MLL aberrations, and deletion of 9p, with conflicting reports on the prognostic relevance of specific abnormalities [6-12]. Furthermore, recent genome wide and exome analyses have revealed that each ETV6/RUNX1 patient could have multiple mutations, underscoring the heterogeneity of this ALL subtype [13,14]. Importantly, this genetic heterogeneity may influence overall treatment response and survival, resulting in differing outcomes for specific patients.
Few studies of children with ETV6/RUNX1 (+) ALL from Korea have been conducted. Whether the outcome of this pediatric ALL subgroup is similar to that reported from other countries, and the important factors that influence outcome are largely unknown. In this study, we reviewed the outcome of children with ETV6/RUNX1 (+) ALL treated at our institution over 10 years and attempted to identify significant prognostic factors with regards to survival.
Materials and Methods
1. Patients
All patients diagnosed with ETV6/RUNX1 (+) ALL from January 2005 to December 2011 at the Department of Pediatrics, The Catholic University of Korea were included in the initial study cohort. Positivity for the cryptic ETV6/RUNX1 rearrangement was confirmed by either fluorescence in situ hybridization (FISH) or reverse-transcriptase polymerase chain reaction screening at diagnosis as previously reported [15]. A chromosomal study of metaphase bone marrow (BM) cells was also conducted for each patient using conventional G-banding analysis to identify cytogenetic abnormalities in addition to the cryptic ETV6/RUNX1 rearrangement.
During the study period, a total of 65 patients were diagnosed with ETV6/RUNX1 (+) ALL. Excluding one patient who was transferred to another institution for further treatment after achieving complete remission (CR), and one patient who received allogeneic hematopoietic cell transplantation (HCT) in first CR due to concurrent hypodiploidy, the final study cohort consisted of 63 patients who received chemotherapy only as first-line treatment (Table 1).
2. Treatment regimen
The major characteristics of our institution’s treatment regimen have been reported previously [16]. Patients were classified as low, standard, high, or very high risk according to institution criteria (Supplementary Table 1). With regards to the chemotherapy regimen, all patients received uniform remission induction and consolidation chemotherapy. Afterwards, low and standard risk patients received one phase each of interim maintenance and delayed intensification, while high and very high risk patients received two phases each of interim maintenance and delayed intensification before maintenance chemotherapy (Supplementary Tables 2-7). During maintenance treatment, all patients received high dose methotrexate at 12 weekly intervals. None of the patients received prophylactic cranial irradiation in first CR. As an adjustment of institution protocol, all patients diagnosed from January 2009 onwards received prednisolone during all phases of steroid treatment (n=31), whereas previously dexamethasone had been administered (n=32).
3. Measurement of minimal residual disease
A BM study was conducted at the end of induction, end of consolidation, prior to each phase of delayed intensification, and at the start and end of maintenance treatment. In these studies, positivity for ETV6/RUNX1 rearrangement was measured using real-time quantitative-polymerase chain reaction (RQ-PCR) and FISH.
The ETV6/RUNX1 fusion transcript was measured by RQ-PCR using the ETV6/RUNX1 Quantification kit (Biosewoom Inc., Seoul, Korea). The samples were assayed on an ABI-Prism 7500/7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The relative expression level of the gene of interest was calculated using the 2–ΔΔCt method by normalizing to the geometrical mean of ABL1 as a stable reference gene and presented relative to the control. FISH testing was conducted using ETV6/RUNX1 translocation, dual fusion probe (Cytocell, Cambridge, UK). Pretreatment and hybridization were performed in accordance with the manufacturer’s recommendation.
Consistent with a previous study that found patients who relapsed had RQ-PCR levels between 10–4 and 10–2 at the end of remission induction [17], patients with RQ-PCR ≥ 1×10–3 (the midpoint of 10–4 and 10–2) at end of induction (day 30, time point 1 [TP1]), or ≥ 1×10–4 at end of consolidation (day 85, time point 2 [TP2]) were considered to have minimal residual disease (MRD [+]). Also, patients with ≥ 0.5% cells containing the ETV6/RUNX1 rearrangement among the 200-400 interphase cells examined using FISH at either TP1 or TP2 were also considered to be positive for MRD. Patients who were MRD (+) at TP1 were classified and treated as high risk, even if all other parameters indicated low or standard risk.
Of note, RQ-PCR measurement of ETV6/RUNX1 was incorporated into our diagnostic methodology in 2007, prior to which only FISH had been used and precluding the use of RQ-PCR in all patients of the cohort. Of the 63 patients in the study cohort, 27 and 29 were evaluated for ETV6/RUNX1 using RQ-PCR at TP1 and TP2, respectively. Also, 60 patients each were tested using FISH at TP1 and TP2. Overall, 62 patients each were evaluated for MRD at TP1 and TP2 using RQ-PCR or FISH.
4. Definition of study parameters and endpoints
The EFS and overall survival (OS) of the study cohort were analyzed. EFS was defined as the time from diagnosis of ALL to the last follow-up in CR or the first event that included relapse, primary refractory disease, death, or secondary malignancy. OS was defined as the time from diagnosis of ALL to last follow-up or death from any cause.
The following factors were analyzed for their impact on EFS: patient sex, age at diagnosis, initial white blood cell (WBC) count, type of steroid used for treatment, response to prephase steroid treatment, risk group according to National Cancer Institute (NCI)/Rome criteria based on age and initial WBC count [18], and presence of MRD at either end of induction or end of consolidation. Because of the reported high incidence of an abnormal karyotype in conjunction with the ETV6/RUNX1 rearrangement in ETV6/RUNX1 (+) ALL patients [6], the presence of a complex karyotype was studied as a cytogenetic variable potentially influencing EFS. Chromosome 12 has been reported to be the most frequently involved additional cytogenetic abnormality, most often manifesting as deletion of the remaining TEL on 12p [6,7]. Hence, deletion of 12p (del(12p)) and structural abnormalities (defined as translocations or inversions, not additions or deletions) involving 12p in addition to ETV6/RUNX1 were also evaluated for prognostic significance. A complex karyotype was defined as a karyotype with three or more structural or numerical abnormalities, as previously described [19].
5. Statistical analysis
Univariate study of impact of clinical and cytogenetic factors on EFS was conducted with the Kaplan-Meier method and comparisons were made with the log-rank test. Collinearity among variables found to be significant in the univariate study was assessed by checking the variance inflation factor (VIF) and the tolerance statistic. A multivariate study was done with Cox proportional hazard regression for variables found significant in the univariate study. Incidence of relapse was calculated with the cumulative incidence function. p-values of < 0.05 were considered significant.
Results
1. Cytogenetic studies
Of the 63 patients in the cohort, a complex karyotype was found in 30 (48%), while deletion of the remaining 12p was observed in 14 patients (22%). Abnormalities involving MLL were found in six patients (10%) (deletion 5, addition 1). Structural abnormalities involving 12p in addition to ETV6/RUNX1, defined as translocations or inversions, were also found in six patients (10%) (Table 2).
2. Response to initial treatment and MRD
All patients achieved CR at the end of 4 weeks of remission induction chemotherapy. Of the 62 patients who were evaluated for MRD at TP1 and TP2, six (10%) and two (3%) patients were found to be MRD (+) at TP1 and TP2, respectively (Table 1). At TP1, four patients were diagnosed as MRD (+) by RQ-PCR, and two patients by FISH. At TP2, two patients were found to be MRD (+) by RQ-PCR.
Of the 27 patients who had MRD measured at TP1 using RQ-PCR, 19 were detected at < 10–4 (70%), four were detected at 10–4, three were detected at 10–3, and one patient was found to be polymerase chain reaction (PCR) positive at 10–2. Of the 29 patients who had MRD measured at TP2 using RQ-PCR, 27 were detected at < 10–4 level (93%) and two were found to be PCR positive at 10–4. Among the four patients who were PCR positive at ≥ 10–3 at TP1, and hence MRD (+) according to the study definition, all but one were detected at < 10–4, and hence MRD (–), at TP2.
Of the 60 patients who were evaluated by FISH at TP1, two were MRD (+) at 2.5% and 0.5% of cells counted. These two patients had not been evaluated by RQ-PCR. None of the patients evaluated by FISH at TP2 were found to have MRD.
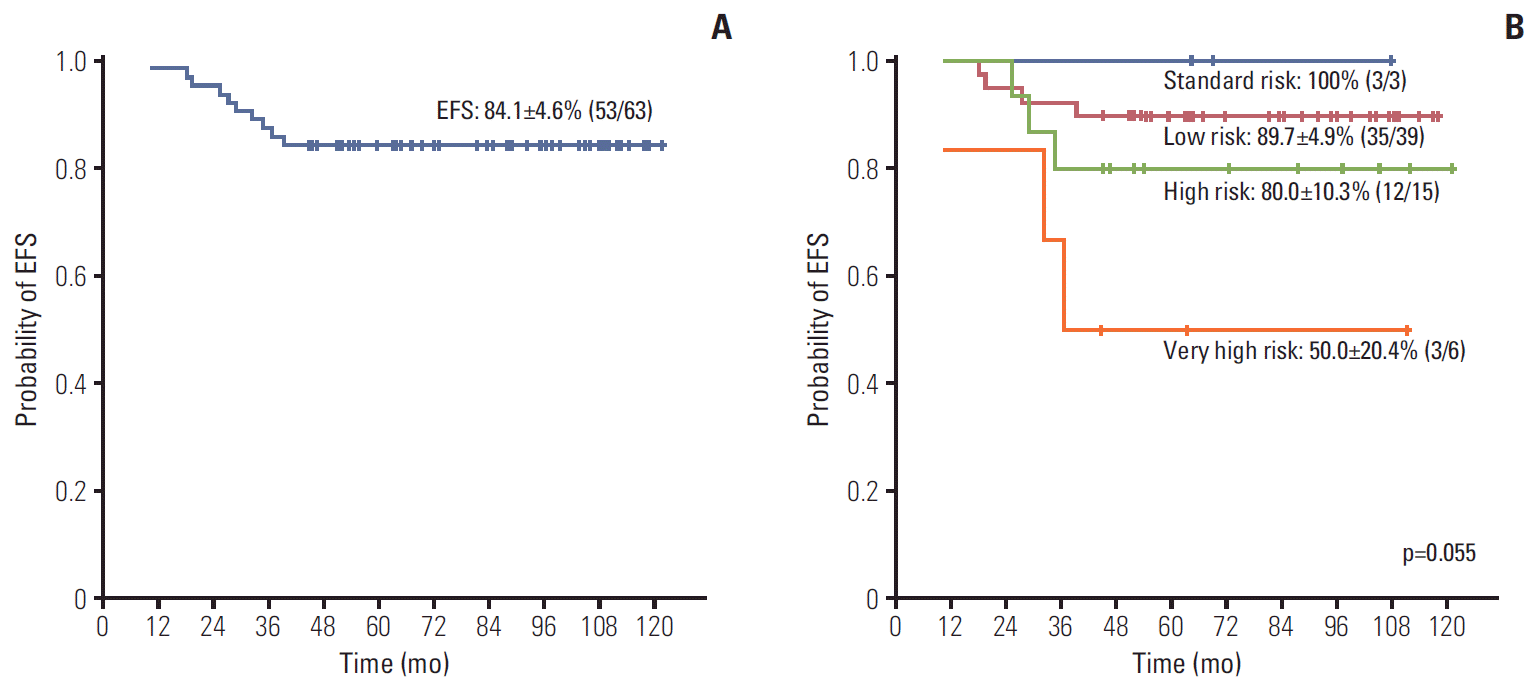
3. Event-free survival
The 5-year EFS for the ETV6/RUNX1 (+) ALL cohort was 84.1±4.6% (53/63) (Fig. 1A). All events were relapses that occurred at a median of 28.3 months from diagnosis (range, 10.4 to 39.4 months). The EFS differed according to overall patient risk group, with low risk patients having 89.7±4.9% EFS and very high risk patients having 50.0±20.4% EFS (p=0.055) (Fig. 1B). Univariate analysis of prognostic factors for EFS revealed that only MRD at TP1 and the presence of additional 12p structural abnormalities had a significant impact, with the presence of either predicting worse outcome (Table 3, Supplementary Fig. S1). Among the four patients who were MRD (+) at TP1 and subsequently relapsed, none were found to be MRD (+) at TP2. Of note, all patients who had MRD < 10–3 at TP1, and were hence MRD (–) according to the study definition, survived event-free. Of the three patients with additional 12p structural abnormalities who relapsed, two died from disease progression.
Multivariate analysis revealed that both MRD at TP1 (hazard ratio [HR], 17.54; 95% confidence interval [CI], 3.83 to 80.29; p < 0.001) and the presence of additional 12p structural abnormalities (HR, 13.06; 95% CI, 2.59 to 65.89; p=0.002) had a significant negative impact on EFS (Table 4). Tests indicated limited collinearity between the two significant variables (average VIF, 1.006; average tolerance, 0.994).
Overall, 10 of 63 patients relapsed, nine in the BM and one with isolated central nervous system (CNS) relapse, resulting in a 5-year cumulative incidence of relapse of 15.9±4.6%. All but one of the patients relapsed prior to completion of planned chemotherapy. None of the patients in the cohort died in remission.
4. Treatment of relapsed patients
All nine patients with BM relapse underwent allogeneic HCT after achieving second CR, four from matched sibling donors, four from unrelated donors, and one from a haploidentical family donor. Eight of nine patients subsequently relapsed at a relatively short period from transplant (median time to relapse, 4.9 months; range, 2.0 to 12.0 months). All eight patients died of disease progression at a median of 11.0 months from HCT (range, 3.8 to 20.0 months). One of the nine patients survives without event 14.2 months after HCT. One patient who showed isolated CNS relapse 32.5 months since diagnosis received craniospinal irradiation and intensified systemic and intrathecal chemotherapy, and survives in second remission 32.2 months since relapse. The 8-year OS of the ETV6/RUNX1 (+) ALL cohort was 82.5±6.9% (55/63).
Discussion
Here, we report the outcome and prognostic factors for ETV6/RUNX1 (+) ALL in Korean children treated at a single institution. The 5-year EFS for our study cohort was 84.1±4.6%, which is lower than results reported previously by other institutions [2,5]. Disparities in outcome between our ETV6/RUNX1 (+) ALL patients and those previously reported also included the time to event and final OS. Relapses occurred at a short median of 28.3 months from diagnosis, with nine of the 10 patients relapsing prior to treatment completion, while the Nordic Society of Pediatric Hematology and Oncology study noted that many of their patients experienced a late relapse [3]. Moreover, previous studies reported an OS greater than 90%, indicating that a significant portion of relapsed patients were cured with further therapy [2,3,5]. One study that focused on relapsed ETV6/RUNX1 (+) ALL patients found a median time to relapse from diagnosis of 42.5 months, and a significantly better OS after relapse for ETV6/RUNX1 (+) patients compared with those who were ETV6/RUNX1 (–) [20]. This experience contrasts with both the shorter time to relapse in our patients, and the poor outcome of our 10 relapsed patients, only two of whom currently survive in remission, despite treatment with allogeneic HCT in nine patients with BM relapse. Although validation from a larger cohort is necessary, our results indicate the possibility of a subgroup of ETV6/RUNX1 (+) ALL with guarded prognosis who may relapse early and may not be cured despite intensive treatment subsequent to relapse. Identifying prognostic factors that define this subgroup may help clarify the heterogeneity of ETV6/RUNX1 (+) ALL patients and improve their overall outcome.
Among variables that influence EFS, we found MRD at TP1, which is at end of remission induction, as detected by RQ-PCR for the ETV6/RUNX1 transcript or by FISH, as well as additional structural abnormalities involving 12p in the karyotype to be significant. Few studies have investigated the role of RQ-PCR measurement of the ETV6/RUNX1 transcript as a means of detecting MRD [21,22]. One study found that four relapsed patients of a cohort of 57 with ETV6/RUNX1 (+) ALL were MRD (+) as detected by RQ-PCR for the ETV6/RUNX1 transcript at the end of remission induction [17]. Three of the four patients had MRD between 10–4 and 10–2, while one relapsed patient had a low level of MRD at < 10–4. In our study, the cut-off level of 10–3 at TP1 significantly predicted EFS. Whether a lower level of MRD at this treatment point better defines a subset of patients with poor prognosis will require a study of a greater number of patients. However, it is important to note that all of the 23 patients with MRD < 10–3 at TP1 in our cohort currently survive event-free. In contrast to the prognostic relevance of MRD at TP1, MRD detected at TP2 failed to have significance.
Additional genetic abnormalities found in the ETV6/RUNX1 (+) ALL blast may also aid in defining patients with poor outcome. Several studies have reported deletion of the non-translocated ETV6 allele as a recurrent abnormality occurring in conjunction with ETV6/RUNX1 [6-9]. In our cohort, del(12p) was found in 14 patients (22%), but was not found to affect EFS. Abnormalities involving 12p have been reported to be the most common additional structural aberrations in ETV6/RUNX1 (+) ALL patients [6]. To verify the impact of these abnormalities separately from del(12p), we narrowly redefined structural abnormalities of 12p as translocations or inversions. Although we only identified six patients with such additional abnormalities, it is important to note that three of these patients relapsed, and two subsequently died from disease progression.
It is important to note that this study has several limitations. RQ-PCR based MRD monitoring was only conducted in a subgroup of the study cohort, with the remaining patients analyzed by FISH, resulting in lack of a uniform method of testing for MRD. Moreover, only 10 patients in the cohort of 63 patients relapsed, giving a cumulative incidence of 15.9%. As ETV6/RUNX1 (+) ALL is known to have favorable outcome, a study of a larger number of patients is required to more accurately validate the prognostic factors, that is MRD at end of remission induction and the presence of additional 12p structural abnormalities, underlined in this study. Finally, our data collection and analysis were limited by the retrospective nature of this study.
Conclusion
In summary, our ETV6/RUNX1 (+) ALL cohort had a 5-year EFS of 84.1%. Relapses occurred relatively early during the course of treatment, and eight of ten relapsed patients died of disease progression, despite therapeutic measures such as allogeneic HCT. Significant MRD at the end of remission induction chemotherapy and the presence of additional structural abnormalities of 12p in the karyotype in the form of translocations or inversions adversely affected EFS. Larger scale studies will be necessary to confirm the factors that characterize a potential subset of ETV6/RUNX1 (+) ALL with poor outcome, and to improve the survival of this heterogeneous subtype of childhood ALL.
 XML Download
XML Download