

 PDF
PDF Citation
Citation Print
Print


Introduction
Esophageal cancer is the eighth most prevalent cancer and the sixth leading cause of cancer deaths throughout the world [1]. At present, the treatments for esophageal cancers include surgical resection, radiotherapy and chemotherapy [2,3]. However, the curative effects of the existing treatments appear to be insufficient, as reflected by the sub-optimal 5-year survival rate (almost less than 20%) and the observed toxicities of the anti-cancer drugs [3,4]. Therefore, identification of new anti-cancer drugs for treatment of esophageal and other cancers has become a crucial issue.
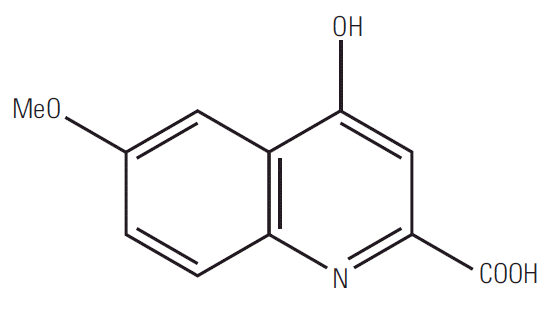
Quinoline derivatives, which have been widely reported to possess a broad range of pharmaceutical activities, can be isolated from different plant sources [5]. The first commonly known natural quinoline compound, 4-hydroxy-6-methoxyquinoline-2-carboxylic acid (Fig. 1), was extracted from Ephedra pachyclada ssp. sinaica, which has been widely used in traditional herbal medicine against allergy, inflammation, microbial and cardiovascular diseases, and cancer [6,7]. 8-Hydroxyquinoline and its derivatives were also reported as natural products such as those isolated from the roots of Centaurea diffusa with phytotoxic activities [8] and Suaeda corniculata with anti-bacterial and anti-fungal activities [9]. Therefore, researchers have further improved the efficacy and potency of quinoline by modifying its structure. Our previous findings also revealed that the 8-hydroxyquinoline derivatives showed relatively promising in vitro and in vivo anti-cancer effects [5], as well as anti-bacterial effects, implying the importance of the 8-hydroxyl group for their biological actions [10].
Peroxisome proliferator-activated receptor (PPAR), a member of the nuclear hormone receptor superfamily, exists as three main subtypes, PPARα, PPARγ, and PPARδ (also known as PPARβ). These receptors can also act as ligand-activated transcription factors. PPARδ is ubiquitously expressed in most human tissues, and many studies have shown that PPARδ is highly overexpressed in tumor cells relative to non-tumor cells. Moreover, it has been shown to be involved in cell differentiation and inflammation, which are highly related to tumor development [11]. In the ligandbound state, PPARδ can bind to an activator and associate with the peroxisome proliferated response element to promote gene transcription. The potential pathways related to the PPARδ for inducing tumor growth include the cyclooxygenase 2 (COX-2) [12,13]. It has also been reported that there are solid correlations among the pathways of PPARδ, overexpression of COX-2 and tumor development [12]. Thus, targeting PPARδ forms a very important direction for exploring novel anti-cancer agents.
Cyclooxygenase, which is also known as prostaglandin H synthase, exists as two main isoforms, COX-1 and COX-2. COX-1 is constitutively expressed in most mammalian cells and has functions such as regulation of renal blood flow and platelet aggregation [14]. However, COX-2 is inducible and usually overexpressed in gastrointestinal cancers, including esophageal cancer [15,16]. Previous studies showed that COX-2 is highly related to tumorigenic events involving cell proliferation, invasion, apoptosis, inflammation and angiogenesis [17,18]. Moreover, it has been shown that COX-2 derived prostaglandin E2 (PGE2) is one of the potential products that promotes development of tumors [19]. PGE2 is a well-known molecule that participates in tumorigenesis by binding to complementary EP2 and EP4 receptors on cell membranes [16].
In this study, a novel quinoline derivative (8-(4-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydro-2-methylquin-oline, named as 83b1) was examined through in vitro and in vivo anti-cancer tests on esophageal cancer cell lines. The associated mechanisms were also investigated based on molecular docking analysis to reveal PPARδ as a target in cancer treatment.
Materials and Methods
1. Reagents and materials
Cell culture medium RPMI-1640, F-12, Dulbecco's modified Eagle's medium, minimum essential medium α (MEMα), keratinocyte serum-free medium (KSFM), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from Life Technologies (Carlsbad, CA). 83b1 was completely dissolved in dimethylsulfoxide (DMSO).
2. Synthesis of 83b1 with electrospray ionization mass spectrometry and 1H-NMR examination
8-(4-(Trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydro-2-methylquinoline (compound 83b1) (Fig. 2) was prepared according to our previously reported method of asymmetric hydrogenation on 8-(4-(trifluoromethyl)-benzyloxy)-2-methylquinoline, which was obtained through nucleophilic substitution of commercially available 8-hydroxy-2-methylquinoline (Sigma-Aldrich, St. Louis, MO) [20]. The structure and purity of 83b1 was examined through 1H-NMR and liquid chromatography mass spectrometry (LC-MS).
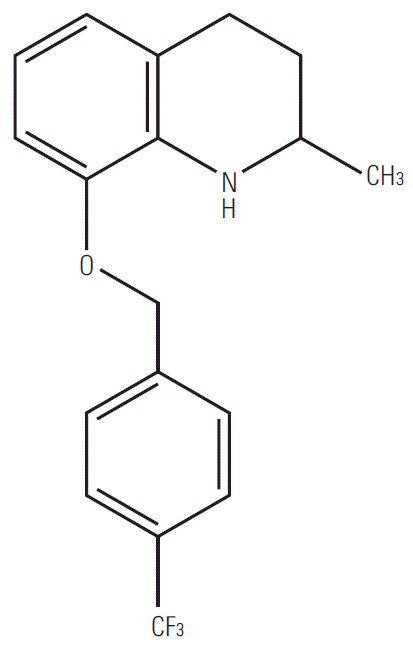
83b1, which appeared as a white solid, was dissolved in CDCl3 completely and examined by 1H-NMR (Supplementary Fig. S1). Upon analysis the following spectrum was reported: 1H-NMR (500 MHz, CDCl3): δ 1.30 (d, 3H, J=6.0 Hz), 1.63-1.72 (m, 1H), 1.95-1.98 (m, 1H), 2.75-2.79 (m, 1H), 2.85-2.90 (m, 1H), 3.39-3.43 (m, 1H), 4.17 (bs, 1H), 5.13 (q, 2H, J=12.5 Hz), 6.54 (t, 1H, J=8.0 Hz), 6.68 (d, 1H, J=8.0 Hz), 6.72 (d, 1H, J=7.5 Hz), 7.55 (d, 2H, J=8.0 Hz), 7.66 (d, 2H, J=8.5 Hz). These findings are consistent with the expected structure shown in Fig. 2. The purity and molecular weight of the synthesized 83b1 were further examined by LC-MS (Supplementary Fig. S2). According to the data obtained, there were three peaks separated in different retention periods. The peaks were analyzed by electrospray ionization mass spectrometry (Supplementary Fig. S3), and the peak comprising 99.7% total area showed a m/z ratio of 83b1 (molecular weight 321 g/mol), whereas the other two peaks comprising 0.3% total area were found to be impurities. Therefore, the purity of 83b1 is very high.
3. Cell lines and culture conditions
One of the esophageal squamous cell carcinoma (ESCC) cell lines of Hong Kong Chinese origin, SLMT-1 [21], was kindly provided by Professor Gopesh Srivastava of the Department of Pathology, the University of Hong Kong. The other two ESCC cell lines of Hong Kong Chinese origin, including HKESC-2 and HKESC-4, and three ESCC cell lines of Japanese origin, including KYSE-150, KYSE-450, and KYSE-520 [22], were purchased from DSMZ (Braunschweig, Germany). Two non-tumor esophageal epithelial cell lines including NE-1 [23] and NE-3 [24] were kindly provided by Professor George S. W. Tsao from the Department of Anatomy, the University of Hong Kong. A human skin cell line, HEK001, was purchased from the American Type Culture Collection (ATCC) and cultured as suggested. The culture medium for KYSE-150 and KYSE-450 was RPMI with 45% F-12 and 10% FBS, that for KYSE-520 was RPMI with 10% FBS, that for SLMT-1, HKESC-2, and HKESC-4 was MEMα with 10% FBS, and NE-1, NE-3, and HEK001 were cultured on KSFM with complementary supplements. All media were supplemented with 100 units/mL penicillin G and 100 μg/mL streptomycin, and all cell lines mentioned above were cultured at 37°C in a humidified incubator with 5% CO2.
4. In vitro cytotoxicity studies by MTS
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed to evaluate the growth inhibitory effects of 83b1 on the six described ESCC cell lines and three immortalized non-tumor cell lines using the CellTiter96 AQueous One Solution (Promega, Madison, WI) for cell proliferation assay as previously reported [25]. In addition, a widely used anticancer drug, cisplatin (CDDP), was used as a positive control [26]. Briefly, 5×103 cells were seeded onto each well of the 96-well plate and allowed to grow for 24 hours. After incubation for 24 hours, different concentrations of 83b1 and CDDP (0, 1.5625, 3.125, 6.25, 12.5, 25, and 50 μg/mL) were added as the treatment. The results were recorded after 72 hours of the treatments using a micro-plate reader to measure the absorbance at 495 nm.
5. Molecular docking analysis
Evaluation of the possible molecular binding targets of 83b1 was conducted based on the similarity ensemble approach (SEA) using the search engine available from http://sea.bkslab.org. The binding of 83b1 to the protein targets was predicted based on molecular structures matched against the ChEMBL medicinal chemistry database version 12 as previous described [27]. Another docking program involving the molecular docking server (http://www.dockingserver.com/web) was used to determine the binding affinity of 83b1 to its predicted target relative to the natural ligand of the target. The docking calculations were conducted using DockingServer [28].
6. Reverse transcription polymerase chain reaction and quantitative real-time polymerase chain reaction
The gene expression levels of COX-2 in the four ESCC cell lines (KYSE-150, KYSE-450, SLMT-1, and HKESC-4) with the treatments of 83b1 were studied by quantitative real-time polymerase chain reaction (qPCR). Approximately 1×106 cells from each cell line were evenly separated into eight replicates and allowed to seed for 24 hours. Three different concentrations of 83b1 (5, 10, and 20 μg/mL) and a negative control (0.05% DMSO) were used to incubate the cells for 24 hours. The total RNA of each the cell line was extracted using a RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Two micrograms of extracted RNA was reverse-transcribed to cDNA using reverse-transcriptase (Promega).
Two-step reverse transcription polymerase chain reaction (RT-PCR) assay reagents with SYBR Green (Promega) were used for the qPCR reactions, which were conducted as follows: pre-denaturation for 2 minutes at 95°C; polymerase chain reaction (PCR) amplification for 40 cycles of 15 seconds at 95°C and 2 minutes at 60°C. The threshold cycle (Ct) was recorded using a Pikoreal real-time PCR system (Thermo Scientific, Waltham, MA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control to normalize the quantity of cDNA synthesized from all cell lines. The specific primers for COX-2 were as follows: 5′-CCA GCA CTT CAC GCA TCA GT-3′ (forward) and 5′-ACG CTG TCT AGC CAG AGT TTC AC-3′ (reverse) [29] and the specific primers for GAPDH were as follows: 5′-AAA TCA AGT GGG GCG ATG CTG-3′ (forward) and 5′-GCA GAG ATG ATG ACC CTT TTG-3′ (reverse) [30].
7. Enzyme-linked immuno-sorbent assay
The effects of 83b1 on the production of PGE2 in the cell lines (KYSE-150, KYSE-450, and SLMT-1) were examined using an enzyme-linked immuno-sorbent assay (ELISA) kit (Cayman, Ann Arbor, MI). Approximately 1×106 cells from each cell line were evenly separated into eight replicates and then seeded for 24 hours. Three different concentrations of 83b1 (5, 10, and 20 μg/mL) and a negative control (0.05% DMSO) were used to incubate the cells for 48 hours. The culture medium was then removed for quantitative determination of PGE2 with reference to the standard curve according to the manufacturer’s instructions.
8. In vivo studies of 83b1 in nude-mice xenograft with KYSE-450
Athymic nude mice 4 weeks of age were purchased from the State Key Laboratory of Chinese Medicine and Molecular Pharmacology (Shenzhen, China), the Hong Kong Polytechnic University. All procedures were approved by the ethics committee. Approximately 5×106 KYSE-450 cells were injected subcutaneously into the mid-dorsal region of eight athymic nude mice. Tumors were allowed to grow without treatment until a volume of 150 mm3 was reached. The ten nude mice were randomly divided into two groups. 83b1 was then injected into five mice intraperitoneally at 10 mg/kg/day, while the rest of the mice were used as the vehicle control. Tumor volumes were measured daily with calipers and calculated as previously described [25].
Results
1. In vitro cytotoxicity studies
The inhibitory effects of 83b1 on the described ESCC cell lines and immortalized non-tumor cell lines were examined by MTS cytotoxicity assay. As shown in Fig. 3A-F, 83b1 showed comparable inhibitory effect on all ESCC cell lines with CDDP in a dose-dependent manner. The inhibitory effects are summarized in terms of MTS50 (50% reduction of MTS signals compared with the vehicle control) in Table 1. In addition, 83b1 showed significantly weaker cytotoxic effects on NE-1, NE-3, and HEK001 than CDDP (Table 1, Fig. 3G-I).
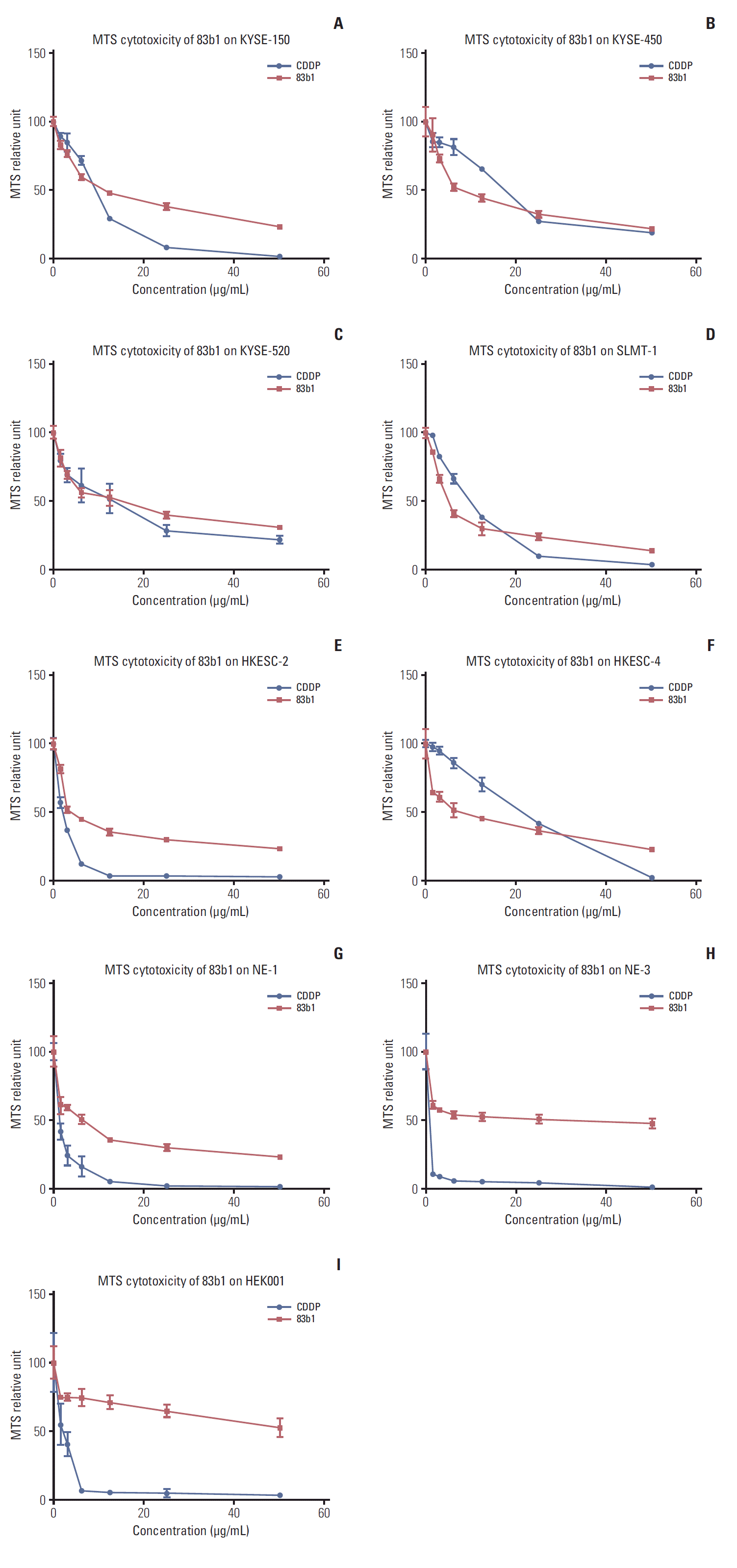
Fig. 3.
(A-I) Inhibitory effects of 83b1 on ESCC and non-tumor cell lines examined by MTS cytotoxicity assay. ESCC, esophageal squamous cell carcinoma; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium.
![]()
Table 1.
Summary of inhibitory effects (MTS50) of 83b1 on ESCC and non-tumor cell lines
![]()
2. Molecular docking analysis
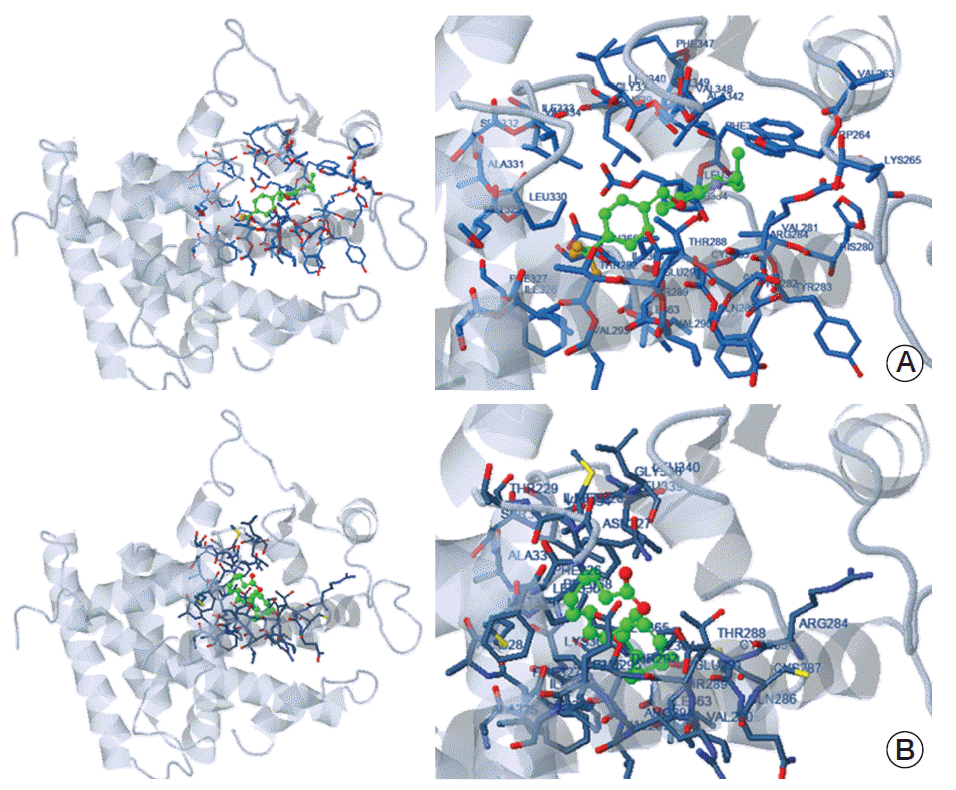
The SEA, which has been shown to be an important step for predicting possible drug target(s), was used to evaluate possible biological targets of 83b1 in cells [26]. The possible predicted protein targets with promising ligand-protein binding are shown in Table 2. These two targets were found to be complementary to 83b1, with the highest complementary score based on their molecular structures according to the ChEMBL medicinal chemistry database indicating that they are protein-tyrosine phosphatase 1C and PPARδ. Another docking program in molecular docking server was utilized to determine the binding affinity of 83b1 to its predicted target, PPARδ (Fig. 4). The free binding energy of 83b1 to PPARδ is –7.41 kcal/mol (Fig. 4A), which shows a more stable and stronger binding than that between PPARδ with its natural ligand, arachidonic acid that the free binding energy of arachidonic acid to PPARδ was estimated to be –5.66 kcal/mol (Fig. 4B).
3. Effects of 83b1 on the mRNA expression of COX-2 in ESCC cell lines
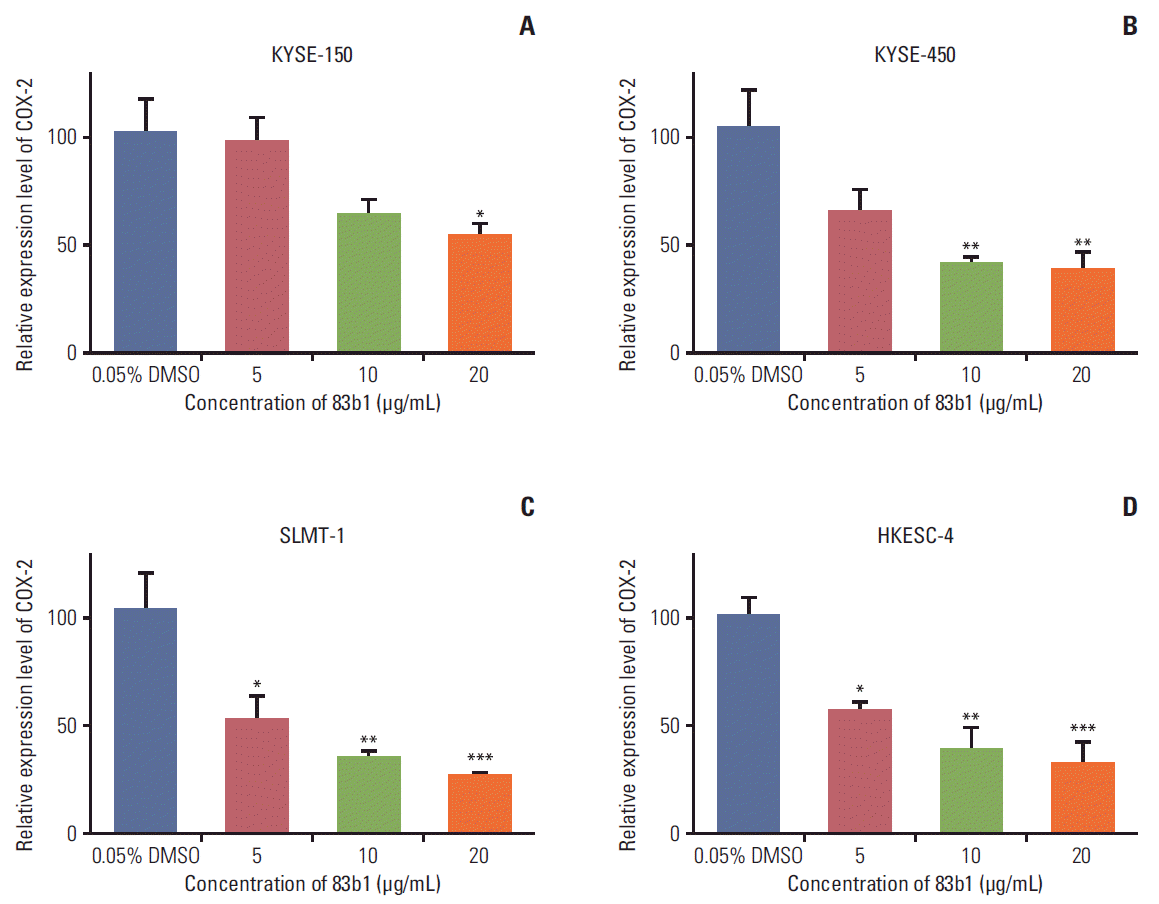
Based on the prediction of PPARδ as possible targets of 83b1 from the molecular docking analysis, qPCR was conducted to determine the down-regulating effect of 83b1 on COX-2 mRNA expression in four of the ESCC cell lines (KYSE-150, KYSE-450, SLMT-1, and HKESC-4). As previously described, the four ESCC cell lines were seeded for 24 hours and three concentrations of 83b1 (5, 10, and 20 μg/mL) were used to treat the cells before harvest. RT-PCR was performed to obtain the cDNA for qPCR to determine the expression of COX-2 mRNA with specific primers. As shown in Fig. 5, the expression of COX-2 mRNA in all four ESCC cell lines was significantly reduced after treatment with 83b1 (p < 0.05, p < 0.01, and p < 0.001 vs. untreated control, n=4).
4. Effects of 83b1 on PGE2 production in ESCC cell lines
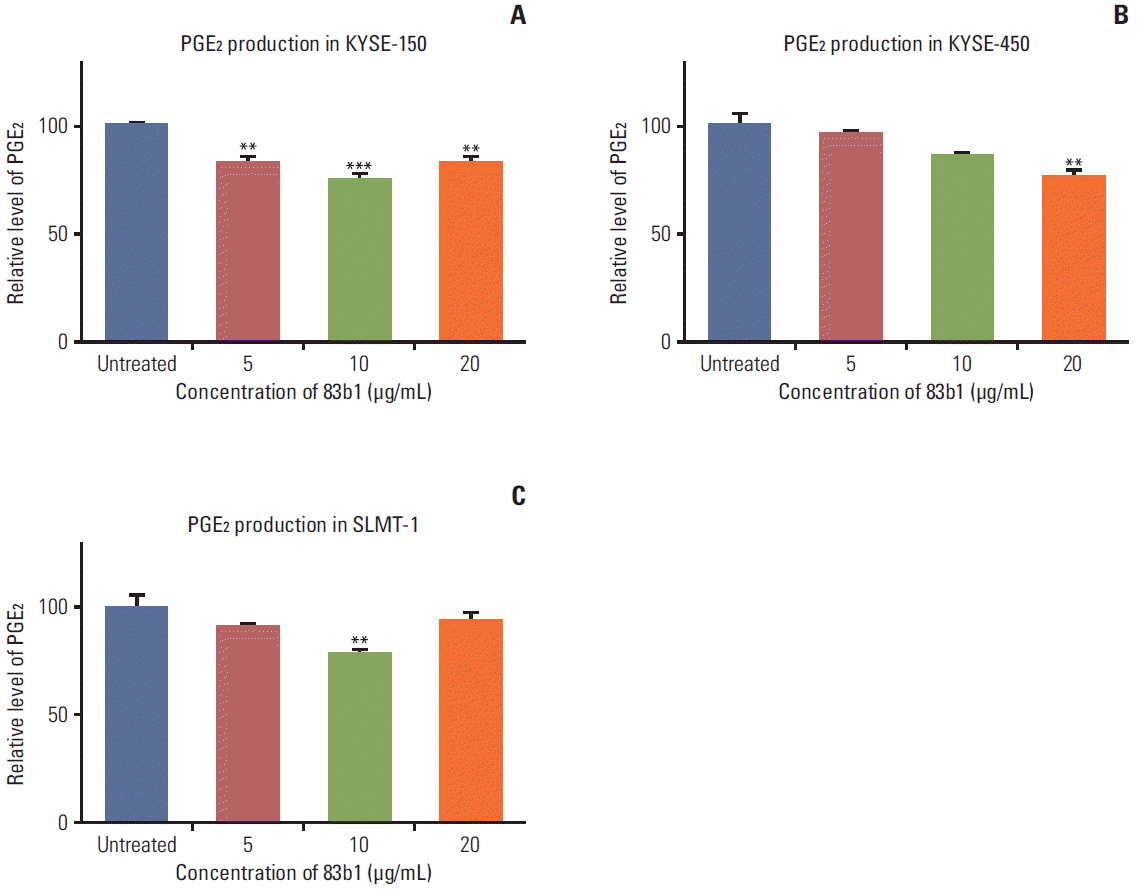
ELISA was performed to estimate the suppressing effects of 83b1 on PGE2 production in the three ESCC cell lines (KYSE-150, KYSE-450, and SLMT-1). As mentioned above, the three ESCC cell lines were seeded for 24 hours, then treated with 83b1 at three different concentrations (5, 10, and 20 μg/mL) for 48 hours before the medium was collected for the assay. As shown in Fig. 6, 83b1 significantly down-regulated the production of PGE2 in all three ESCC cell lines at the respective doses (p < 0.01, p < 0.001 vs. untreated control, n=4).
5. In vivo anti-tumor effect of 83b1 on nude-mice xenograft with KYSE-450
Athymic nude mice xenografted with the human KYSE-450 ESCC cell line were used to test the in vivo anti-tumor effect of 83b1.
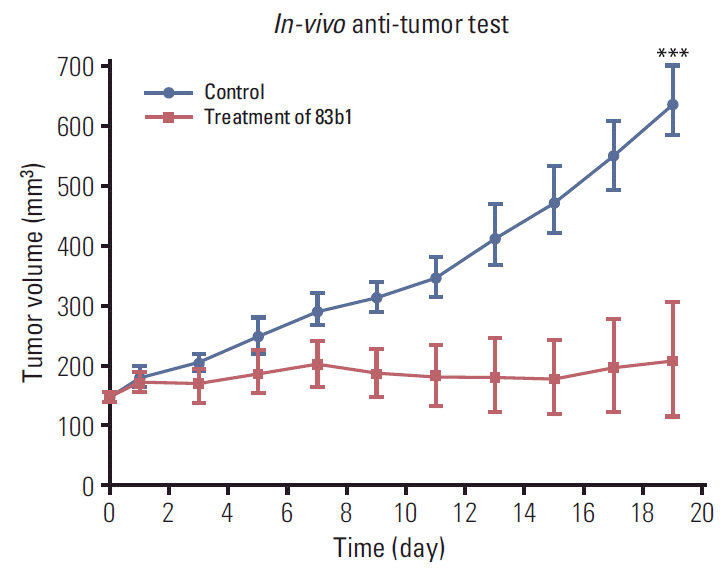
As shown in Fig. 7, 83b1 significantly inhibited the tumor growth in mice when administered at 10 mg/kg/day relative to the vehicle controls. On day 19, the xenografts of the nude mice showed a significant reduction in tumor size after daily treatment with 83b1. Overall, the results showed that 83b1 effectively suppressed tumor growth in vivo (p < 0.001 vs. control, n=5).
Discussion
Natural compounds have been shown to have great potential in pharmaceutical applications. The greatest advantage of these compounds over traditional anti-cancer medicines is that they show much weaker cytotoxicity against normal cells because of their target-specificity. Actually, most existing anti-cancer drugs show very strong inhibitory effects against different kinds of tumors, but also trigger a series of side effects, some of which might be severe. Therefore, chemically modified natural compounds show great potential for use in disease therapies. Chemically modified quinoline-derivatives have wide pharmaceutical power as therapeutic agents, and 83b1 is a quinoline-derivative that has been shown to exert significant anti-tumor effects against ESCC with low cytotoxicity toward non-tumor cells.
In this study, 83b1 showed significantly greater anti-tumor effects against a series of ESCC cell lines relative to the widely used anti-cancer drug, CDDP, with much lower cytotoxicity against nontumor cells. It is an important criterion that anti-cancer drugs are able to suppress cancer growth selectively while they are not provoking damaging effects on normal cells.
Possible targets of 83b1 were determined by molecular docking analysis, which revealed that it has a high binding preference to compete with the natural ligand to target PPARδ, which has been known as an oncoprotein. The functions of PPARδ in COX-2 and COX-2–derived PGE2 production have been described, and the involvement of COX-2 and PGE2 in tumorigenesis in the ESCCs has been widely reported in many studies. Accordingly, it is important to see the effects of 83b1 on the production of COX-2 and COX2–derived PGE2.
Our results showed that 83b1 can significantly down-regulate COX-2 mRNA expression in ESCC cell lines. Moreover, the production of COX-2–derived PGE2 in ESCC cell lines was also significantly reduced by treatment with 83b1. Therefore, it is possible that 83b1 can antagonize its possible target PPARδ and hence suppress cancer growth through down-regulation of COX-2 and PGE2 production. The in vivo study also showed that 83b1 can significantly suppress tumors in animals within 19 days, and that tumors inside of nude mice almost disappeared.
Overall, this study comprehensively described the functions and effects of 83b1 on ESCC cell lines. The findings of this study also revealed the potential use of chemically modified natural compounds in disease therapy.
Conclusion
Chemically modified quinoline derivatives were recently shown to inhibit tumor growth through different kinds of mechanisms by interacting with various targets [5,10]. Here, we demonstrated that a novel quinoline derivative, 83b1, has strong inhibitory effects on tumor growth with less toxicity in non-tumor cell lines and nude mice xenograft models. The predicted target PPARδ of 83b1 has been widely reported as a cancer-promoting protein ubiquitously overexpressed in different types of cancer tissues [26]. We found that 83b1 can target PPARδ, resulting in downregulation of COX-2 mRNA expression and reduced production of PGE2. The results presented herein will greatly advance development of novel drugs for cancer therapy by targeting PPARδ.
 XML Download
XML Download