

 PDF
PDF Citation
Citation Print
Print


Introduction
Progressive multifocal leukoencephalopathy (PML) is a rare and often fatal demyelinating disease of the central nervous system (CNS) that is nearly exclusively seen in immunocompromised patients. PML is caused by reactivation of JC virus (JCV), which subsequently invades oligodendrocytes and astrocytes, thereby leading to their irreparable degeneration [1,2].
The term PML was first mentioned in 1958 to describe a hitherto unknown demyelinating disease in patients with chronic lymphocytic leukemia (CLL) or Hodgkin’s lymphoma [3]. In 1971, the triggering agent of PML was isolated from a patient named John Cunningham, identified as a polyomavirus and named JCV after the patient [4]. In the first decades after its initial description, PML was mainly observed in patients immunocompromised by hematological malignancies and/or treatment with immunosuppressive drugs. The epidemiology of PML was dramatically changed by the emergence of acquired immune deficiency syndrome (AIDS). Indeed, up to 5% of all AIDS patients developed the disease, accounting for about 80% of all PML cases. In the following decades, the introduction of highly active antiretroviral therapy regimens led to a remarkable decrease in its incidence as well as mortality [1,4].
However, PML moved back into focus after introduction of therapeutic approaches based on monoclonal antibodies. Soon after its introduction in 2004, first cases of patients developing PML after treatment with the alpha-4 integrin antibody natalizumab for multiple sclerosis or Crohn disease were reported [2,4]. To date, the overall PML risk for patients on natalizumab was estimated to be 4 to 11 per 1,000 depending on anti-JCV antibody status, treatment duration and prior history of immunosuppression [5]. In the following years, other monoclonal antibodies have also been suspected to increase the incidence of PML. Among these, the CD20 antibody rituximab is playing an important role because of its widespread use in different B-cell lymphomas, and a remarkable number of PML cases have been reported after rituximab treatment [4,6].
Although rituximab-based immunochemotherapy remains the standard of first-line treatment for B-cell lymphomas such as CLL, the importance of small molecules is steadily increasing, especially in patients with refractory or relapsed disease and in those whose initial therapeutic approach is limited by toxicities. One group of these small molecules consists of inhibitors of Bruton’s tyrosine kinase (BTK), a key enzyme in the signaling pathway downstream of the B cell receptor (BCR). Following antigen-dependent triggering of the BCR, BTK is activated, subsequently leading to inhibition of B-cell apoptosis and promotion of B-cell proliferation [7].
Ibrutinib, formerly known as PCI-32765, is an orally bioavailable selective BTK inhibitor that is administered once-daily to prevent B-cell differentiation, proliferation, and survival [7]. In different studies, ibrutinib was highly active in different B-cell lymphomas such as CLL or mantle cell lymphoma combined with a modest toxicity profile [8]. However, few studies have investigated the possible long-term side effects of ibrutinib.
Here, we report the first case of a patient with CLL who developed PML after receiving ibrutinib therapy. To the best of our knowledge, this is the first study to report such findings.
Case Report
1. Initial diagnosis and treatment of CLL
The reported human immunodeficiency virus–negative male patient initially presented in 2005 at the age of 65 years with moderate lymphadenopathy of the cervical, axillary, abdominal, and inguinal lymph nodes, with no hepatomegaly or splenomegaly. The hemogram revealed lymphocytosis without anemia or thrombocytopenia (Binet stage B). A consecutively conducted bone marrow examination revealed 25% infiltration by CLL of B-cell origin without CD20 expression. After observation for over 3 years, the patient showed a general disease progression in November of 2008. At that time, bone marrow examination showed 70% CLL infiltration of the marrow with partially strong and partially weak CD20 expression. Subsequently, therapy with a total of five cycles fludarabine and cyclophosphamide was initiated in private practice. According to the International Workshop Group on CLL (IWCLL) response criteria, complete remission with incomplete bone marrow recovery (CRi) was achieved after three cycles of this treatment regimen.
2. Rituximab-based and further treatment
In February 2012, a significant progression of lymph node manifestations was observed and immunochemotherapy with rituximab and bendamustine was initiated. Two months later, this treatment had to be discontinued because of a newly diagnosed mixed axonal and demyelinating sensorimotor polyneuropathy resulting in hypoesthesia of the trunk, paresthesia of the fingers, slight tetraparesis, mild ptosis, and mild dysarthria. At that time, CRi was achieved. Disease stability was observed during the following months until a recurrent progression in February 2013. Subsequently, a subcutaneous venous port catheter was surgically implanted and the patient was treated with six cycles of cyclophosphamide, epirubicin, and prednisolone. After a phase of stable disease parameters, CLL progressed again in September 2013 with signs of Coombs-positive hemolysis. Three consecutively administered cycles of fludarabine monotherapy had to be substituted by cyclophosphamide because of the insufficient initial response to treatment. However, this regimen had to be discontinued after three cycles because of a beginning bone marrow failure. At that time, a stable disease (according to IWCLL response criteria) was achieved and persisted for more than 1 year.
3. Treatment with ibrutinib
In March 2015, a new disease progression manifesting with lymphadenopathy and hemolysis was treated by initiation of monotherapy with ibrutinib (420 mg daily), which was initially well tolerated by the patient. However, he had to be admitted to our hospital 3 weeks later with fever caused by bilateral pneumonia; therefore, ibrutinib therapy was suspended after a total treatment period of 27 days. The initial antibiotic regimen consisted of piperacillin-tazobactam and clarithromycin and was supplemented by therapeutic doses of co-trimoxazole because of suspected Pneumocystis pneumonia. A consecutively performed bronchoalveolar lavage was positive for Escherichia coli, Candida albicans, Candida krusei, and herpes simplex virus. Hence, the therapeutic regimen was shifted to ceftriaxone, caspofungin, and therapeutic doses of acyclovir, resulting in rapid cessation of fever. During hospital stay, chlorambucil was administered intermittently to reduce leukocyte counts and ibrutinib therapy was not resumed again. After recovery, the patient was discharged to a nursing facility for one week and was able to return home afterwards.
4. Diagnosis and treatment of PML
In May of 2015, the patient was again admitted to our hospital with fever and radiographic evidence of interstitial pneumonia. Because of sustained fever, initial empiric antibiotic therapy with piperacillin-tazobactam and clarclarithromycin was replaced by meropenem and vancomycin and intravenous immunoglobulins were administered to substitute for a lack of immunoglobulins (IgG, 387 mg/dL). Furthermore, the prophylactic antimycotic regimen was replaced by caspofungin according to resistogram after detection of Saccharomyces cerevisiae in the sputum. Subsequently, fever resolved rapidly and regression of pulmonary infiltration was demonstrated by chest radiography over the course of the next few days.
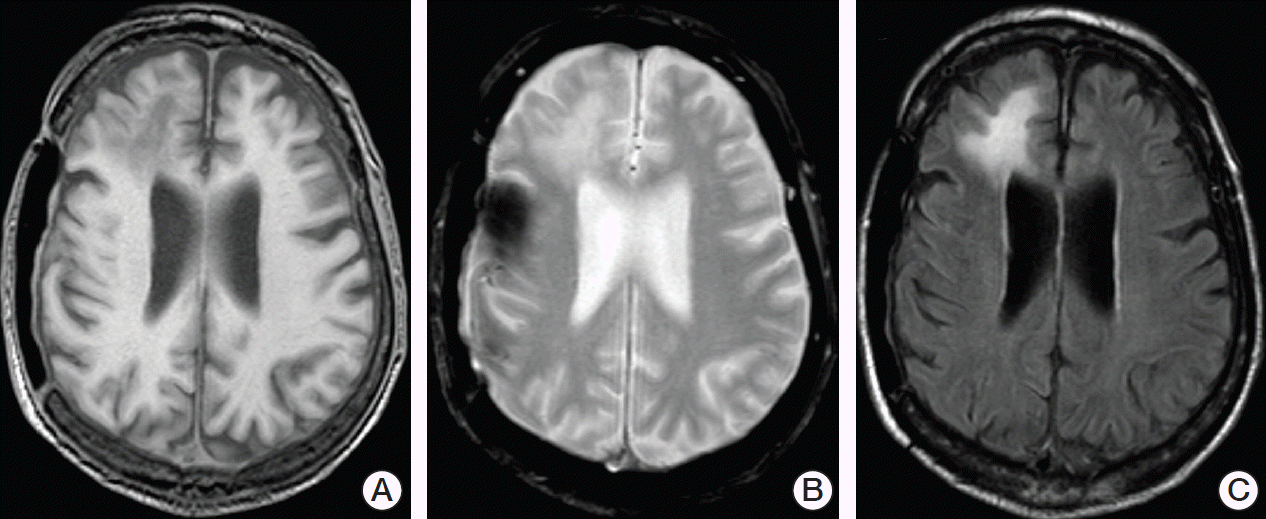
However, additional newly diagnosed and rapidly progressive neurological symptoms consisting of confusion, gait disorder and urinary incontinence were noted during the patient’s hospital stay. An initial cranial computed tomography (CT) scan demonstrated hypodense lesions in the right frontal lobe that were interpreted to be of cicatricial nature. A consecutively conducted lumbar puncture revealed normal cytological and neurochemical parameters of cerebrospinal fluid (CSF) and no increased intracranial pressure (12 cmH2O). Two days later, cranial magnetic resonance (MR) imaging showed the known lesions in the right frontal lobe as hypointense in T1-weighted images and hyperintense in T2-weighted and fluid-attenuated inversion recovery (FLAIR) images without mass effect and with no enhancement of contrast media (Fig. 1). Subsequently, a second lumbar puncture performed because of strong suspicion of PML revealed a highly positive JCV DNA (1.5×107 copies/mL). All other cytological, neurochemical, microbiological and virological tests were normal and PML was diagnosed.
After notification of the diagnosis and prognosis of PML, the patient was discharged on his own and his family’s request with a combined oral therapy regimen with mefloquine (250 mg daily for three days followed by 250 mg once weekly) and mirtazapine (60 mg daily) as proposed in the literature [9]. However, neurological symptoms progressed continuously over the following weeks and the patient died seven weeks after diagnosis of PML at the age of 75 years in a nursing home, most likely due to aspiration pneumonia.
Discussion
Here, we report the eventually fatal case of a 75-year-old male patient with CLL who was pretreated with rituximab and developed PML after receiving monotherapy with ibrutinib, a novel BTK inhibitor. The patient initially presented with typical clinical signs of PML, including speech disorders, cognitive impairment and motor symptoms evolving over days to weeks. Precise neurological symptoms experienced by patients depend on the site of cerebral lesions caused by PML. Therefore, mild gait disorders are likely, as are epileptic seizures [1].
Initial suspicion of PML is often established by cranial imaging performed after appearance of the first neurological symptoms. When compared to CT, MR imaging demonstrates greater sensitivity for visualization of single lesions of PML in the brain and is therefore considered the technique of choice [2]. Cerebral lesions are typically located in both hemispheres in an asymmetric manner, preferably involving subcortical and periventricular white matter in the frontal or parietooccipital lobes. However, involvement of cortical areas has also been reported. Single lesions vary in size and shape, generally becoming larger and more confluent during PML progression. While PML lesions appear hypoattenuating on CT scans, MR imaging shows them as hypointense in T1-weighted images and hyperintense in T2-weighted and FLAIR images. Usually, no mass effect and no enhancement of contrast media is observed. Since signal changes in T1- and T2-weighted MR images are irreversible in most PML cases, diffusion-weighted MR imaging (DW-MRI) constitutes a useful tool to monitor the course of PML. Cytotoxic edema associated with disease progression results in DW-MRI hyperintensity, while quiescent disease areas lead to low signals on DW-MRI [1,2]. Although brain biopsy remains the gold standard to definitively diagnose PML, most cases are diagnosed via polymerase chain reaction (PCR)–based detection of JCV DNA in the CSF. This was also the case in our patient, who was found to have 1.5×107 copies/mL. However, it is important to note that cases with negative JCV PCR results in CSF samples have been reported, despite biopsy-confirmed diagnosis of PML [4].
At present, there is no specific agent to treat PML in a satisfactory manner. To date, no approaches to introduce antiviral drugs to the treatment of PML have demonstrated reasonable efficacy. While cytarabine resulted in decreased replication and multiplication of JCV in a cell culture system, its intravenous or intrathecal application in AIDS patients with PML failed to demonstrate survival benefits in a clinical trial [10]. Different case reports described a possible efficacy of cidofovir, but a prospective pilot study of cidofovir in AIDS patients with PML showed no benefit in MR imaging abnormalities and neurological examination scores [11]. Since the JCV invasion of oligodendrocytes is potentially mediated by binding to the serotonergic 5-HT2α receptor, serotonin receptor antagonists such as the antidepressant mirtazapine or the atypical antipsychotic risperidone have been proposed to block JCV from entering glial cells. However, the results of various case reports are inconsistent, and evidence of efficacy remains lacking [9]. The employment of mefloquine, an antimalarial drug, has been proposed for treatment in response to an in vitro infection assay screening about 2,000 agents for their activity against JCV because it was effective against JCV and showed sufficient penetration into the CNS [12]. A small number of case reports showed mitigated PML courses under mefloquine treatment [9]. However, a randomized proof-of-concept study comparing patients receiving standard care to those receiving additional mefloquine therapy failed to show significant differences in clinical or MR imaging results as well as in JCV DNA loads in the CSF [12]. In our case, a combined regimen of mirtazapine (60 mg daily) and mefloquine (250 mg daily for 3 days followed by 250 mg once weekly) was initiated after initial diagnosis of PML, but was not effective to stop disease progression, leading to fatal outcome seven weeks after diagnosis. Recently, employment of maraviroc, a small molecule inhibitor of chemokine receptor 5, has been discussed to block JCV entry and spread [13]. However, only a small number of cases have been reported and further clinical studies are warranted. To date, reconstitution of the patient’s immune system remains the only option confirmed to successfully treat development of PML [1,2].
Although individual patients with late onset of PML after rituximab treatment have been reported [14], most cases described in the literature occurred within 6 to 12 months after the last application of rituximab [6]. Since our patient received his last rituximab application over 36 months prior to diagnosis of PML, it seems unlikely that rituximab was the only cause of PML development in this case. The same is true for fludarabine. Development of PML after fludarabine treatment have been described in a limited number of case reports, and nearly always in combination with rituximab [15]. Additionally, fludarabine was discontinued about 15 months before PML diagnosis in our case. Therefore, treatment with the novel BTK inhibitor ibrutinib introduced several weeks before the appearance of first clinical PML symptoms and lasting for nearly 1 month must be taken into account. Inhibition of BTK-dependent pathways leads to strong repression of B-cell proliferation and survival [8]. As B-cell–mediated immunity is known to play an important role in control of JCV replication via humoral immune responses, as well as its impact on T-cell activity [1,4], it is conceivable that the introduction of ibrutinib could have triggered JCV reactivation in this case.
This report is intended to increase awareness of this potential side effect. Therefore, risk stratification for development of PML via evaluation of JCV antibody status should be recommended prior to introduction of treatment with ibrutinib. However, further studies are warranted to evaluate the possible association of ibrutinib therapy with development of PML.
 XML Download
XML Download