

 PDF
PDF Citation
Citation Print
Print


Introduction
Compared with standard chemotherapy, significant improvement in response rate and progression-free survival (PFS) has been reported for epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitors (TKIs) including gefitinib, erlotinib, and afatinib in patients with EGFR-mutant non-small cell lung cancer (NSCLC) [1-3]. EGFR-TKIs are now the treatment of choice for patients with EGFR-mutant NSCLC. However, despite the initial response to EGFR-TKIs, most patients develop resistance and finally relapse. Among several mechanisms responsible for acquired resistance (AR) to EGFR-TKIs, the secondary EGFR T790M point mutation in exon 20 is the most common, occurring in approximately 60% of patients. Other mechanisms include MET, HER2, or EGFR amplification; histologic transformation to small cell lung cancer [4]; epithelial-to-mesenchymal transition signature [5]; and AXL kinase activation [6]. Coexistence of other mutations including PIK3CA, HER2, or BRAF mutations also contributes to the development of AR to EGFR-TKIs [7,8].
Poziotinib (NOV120101) is an oral, irreversible inhibitor of EGFR, HER2, and HER4. In preclinical studies conducted in cell lines and xenograft models of NSCLC, poziotinib showed more potent activity than gefitinib, erlotinib, and even afatinib in lung cancer models with EGFR mutations including T790M mutation [9]. In a phase I study to examine the safety and maximum tolerated dose (MTD) of continuous daily dosing of poziotinib in genetically unselected patients with advanced solid cancers including NSCLC, 20% of patients (4/20) experienced partial response (PR), with an MTD of 18 mg and an acceptable toxicity profile, supporting further clinical development of poziotinib. The recommended phase II dose was 16 mg/day [10]. This phase II open-label, single-arm study was conducted to examine the anticancer activity and safety of poziotinib in patients with advanced or metastatic lung adenocarcinoma with activating EGFR mutations, who developed AR to EGFR-TKIs based on the Jackman criteria [11].
Materials and Methods
1. Patients
This phase II, open-label, single-arm study enrolled patients aged ≥ 20 years with histopathologically confirmed stage IIIB or IV lung adenocarcinoma from five institutions in Korea. Eligible patients had at least one measurable lesion or, if not measurable, an evaluable lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST) ver. 1.1 and documented activating EGFR mutations. Patients had received erlotinib or gefitinib as first-line or subsequentline therapy from which they achieved a best overall response of complete response (CR), PR, or stable disease (SD; at least ≥ 6 months) and progressed within the last 30 days. Patients had an Eastern Cooperative Oncology Group performance status of 0-2 with a life expectancy of ≥ 12 weeks; consented to providing tumor tissue samples; and had a white blood cell count of ≥ 4,000/mm3, platelet count ≥ 100,000/mm3, serum creatinine and total bilirubin level ≤ 1.5 times the upper limit of normal (ULN), and serum aspartate aminotransferase and alanine aminotransferase level ≤ 2.5 times the ULN.
The exclusion criteria were as follows: patients with unresolved adverse events (AEs) from erlotinib or gefitinib (Common Terminology Criteria for Adverse Events [CTCAE] grade ≥ 2); resistance to erlotinib or gefitinib secondary to transformation to small-cell lung cancer; major surgery or anticancer therapy other than gefitinib and erlotinib within 4 weeks of the start of the study treatment; untreated symptomatic brain metastasis; interstitial lung disease; active infection; cardiovascular disease or condition including New York Heart Association class III or IV heart failure, uncontrolled hypertension, unstable angina pectoris, or myocardial infarction within 6 months of the study start, uncontrolled cardiac arrhythmia, or other clinically relevant abnormalities; resting left ventricular ejection fraction (LVEF) less than the lower normal limit defined by the institution; any gastrointestinal disease or condition having the prominent symptom of diarrhea; a history of malignancy except for treated non-melanomatous skin cancer, in situ cervix carcinoma, or ductal in situ breast carcinoma; and patients who were pregnant, lactating, or using inadequate contraception.
2. Treatment and procedures
Poziotinib was administered orally, consecutively, and once daily in 28-day cycles with a starting dose of 16 mg until progressive disease (PD) or intolerable AEs. For patients who experienced drug-related AEs, treatment with poziotinib was interrupted until the AEs were resolved to CTCAE grade ≤ 1 or baseline level. The treatment was resumed at the previous dose or a reduced dose by 4 mg, and if a dose reduction below 8 mg consecutively once daily was required, the regimen was changed to intermittent dosing (i.e., 2 weeks of 8-mg poziotinib treatment followed by 1 week off treatment). Further dose reduction or up-titration after the reduction was not allowed. Treatment with poziotinib was discontinued permanently in patients requiring a dose reduction below 8-mg intermittent dosing, had not recovered to AEs of CTCAE grade ≤ 1 or baseline level within 2 weeks, had pneumonia or LVEF reduction of CTCAE grade ≥ 3, or had interstitial lung disease or pneumonitis of any grade. Supportive care including anti-diarrhea medication, antibiotics, analgesics, and antiemetics as well as treatments for diseases other than lung adenocarcinoma was allowed, and local palliative radiotherapy to improve symptoms such as bone pain, pulmonary occlusion, and skin lesions was also allowed.
Tumors were assessed by spiral computed tomography or magnetic resonance imaging (except for the chest) at baseline (imaging at the time PD from previous EGFR-TKIs was confirmed and could be used if obtained within 2 weeks prior to baseline), week 4, week 8, and every 8 weeks thereafter until PD or the start of new anticancer therapy. After discontinuation of treatment, patients were followed-up every 3 months until death or the start of new anticancer therapy. Scans were reviewed both locally and centrally. Tumor response assessments were based on RECIST ver. 1.1. Tumor tissue samples were mandatory at screening from all patients enrolled (except for those with available tumor tissue collected within 2 weeks of the study start) and optional after PD. Blood samples for genotyping were obtained from patients at screening, cycle 3, and every two cycles thereafter until the end of treatment. Safety assessments were performed at least every cycle using CTCAE ver. 4.0.
The study was approved by the ethics committee of each study institution and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines, and all patients provided written informed consent.
3. Study outcomes
The primary endpoint of the study was PFS by independent review. The secondary endpoints included PFS rate at week 16, objective response rate (ORR; CR+PR), disease control rate (DCR; objective response+SD), overall survival (OS), duration of objective response, and duration of disease control. PFS was calculated from the start of treatment to PD or death, whichever occurred first, or was censored at the last imaging date in the absence of PD or death. OS was calculated from the start of treatment to death or was censored at the last date of contact if the patient was alive. Safety assessments included treatment-related AEs, laboratory tests, vital signs, radiography, electrocardiogram (ECG), and LVEF by a multi-gated acquisition scan or echocardiogram.
4. Prestudy tumor tissue analysis
DNA extraction and mutation analysis of prestudy tumor samples were performed in a central laboratory (Theragen Etex, Suwon, Korea). Mutational status was analyzed by Ion Torrent deep-amplicon sequencing using an Ion Torrent AmpliSeq Cancer Hotspot customized lung cancer panel (Thermo Fisher Scientific Inc., Waltham, MA). Fluorescent in situ hybridization (FISH) of MET (c-MET Probe KBI-10719, Kreatech, LG Amsterdam, Netherlands), EGFR, and HER2 was performed on available tumor tissues using the standard protocol [7,12], and MET expression status was also analyzed using immunohistochemistry (IHC) (clone SP44, Ventana, Tucson, AZ). Tumors with a staining-intensity score of 3 and with more than 50% positive nuclei were considered to have MET overexpression.
5. Plasma sample analysis
Circulating cell-free DNA was extracted from 200 μL of plasma samples using the QIAamp MinElute virus spin kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The purity and concentration of extracted DNA were determined by spectrophotometry (NanoDrop 2000, Thermo Fisher Scientific Inc.). DNA samples with absorption ratios of 260/280 nm greater than 1.8 were used for subsequent analyses. For plasma samples, only T790M mutation was analyzed using the PANAMutyper R EGFR kit (Panagene, Daejeon, Korea), a new highly sensitive mutation detection kit that uses a peptide nucleic acid clamping–assisted fluorescence melting curve analysis method in mutation detection and genotyping.
6. Statistical analysis
The sample size (n=40) was selected to provide at least 34 patients with PFS event considering 15% of follow-up loss with the following assumption: in a 12-month period of patient enrollment, a total of 34 PFS events in the study would have a 90% power for detection of a 1.8-month improvement of median PFS with poziotinib versus historical control where the conventional anticancer chemotherapies had been implemented (median PFS, 4.5 months vs. 2.7 months for poziotinib and historical control [13,14], respectively), with a one-sided significance level of 5%. The primary endpoint, median PFS, and the secondary endpoints including median OS, time to progression, time to objective response, duration of objective response, and duration of disease control were calculated from Kaplan-Meier estimates with 95% confidence interval (CI). For other secondary endpoints including PFS rate at 16 weeks, ORR, and DCR, the number and proportion of patients was calculated with 95% CIs. Statistical analysis for subgroups based on mutational status was performed using log-rank, Fisher exact, or Pearson’s chi-square tests depending on the types of variables. Efficacy assessments were performed in the full analysis set consisting of patients who received at least one dose of poziotinib with at least one evaluation of the primary efficacy endpoint after baseline. Safety data was summarized descriptively in patients who received at least one dose of poziotinib.
Results
1. Patient characteristics and disposition
Forty patients were enrolled between December 2012 and September 2013. Thirty-nine patients received at least one dose of poziotinib with at least one tumor assessment after baseline and were included in the full analysis set and safety set. One patient who did not receive the study drug was withdrawn from the study and excluded from the data analysis. Demographics and baseline characteristics of patients are shown in Table 1. The median age was 62 years (range, 43 to 84 years), and the majority of patients were women (74%) and never-smokers (77%). All patients had stage IV disease. Most patients received gefitinib or erlotinib as first-line (69%) or second-line (28%) therapy with a median treatment duration of 10.4 months (range, 3.4 to 33.2 months). The best response to previous EGFR-TKIs was predominantly PR (92%). At the time of data cut-off (September 1, 2014), all patients in the full analysis set, except for one who achieved PR with poziotinib, discontinued treatment for the following reasons: lack of efficacy or PD (n=28), voluntary withdrawal of consent (n=7), AEs (n=2), or other reason (n=1).
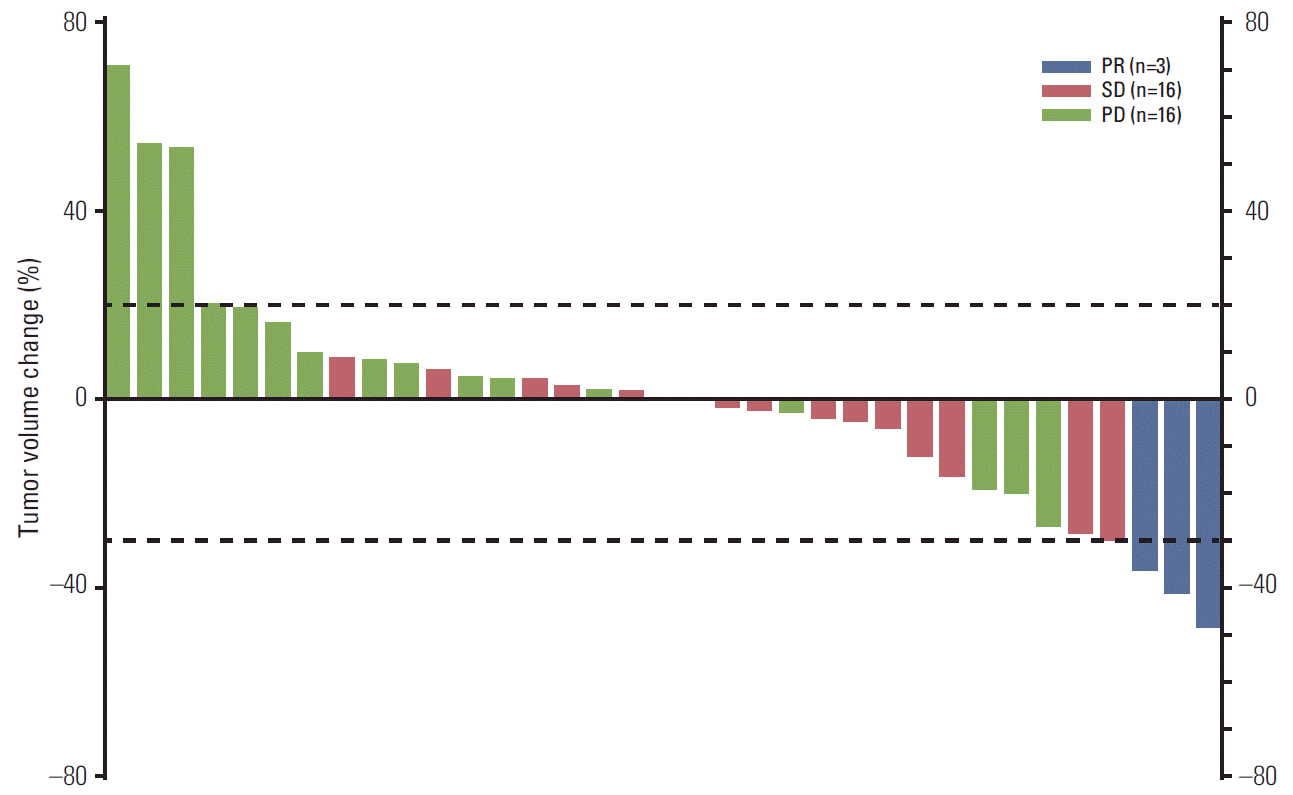
2. Efficacy assessments
A summary of the efficacy measures for poziotinib by independent review is shown in Table 2. The median PFS at the time of analysis (January 7, 2015) was 2.7 months (95% CI, 1.8 to 3.7), and the PFS rate was 35% at week 16 (95% CI, 20 to 51). The best response changes from baseline in tumor lesions were evaluated in 35 patients with measurable lesions at baseline (Fig. 1). Three patients achieved PR (ORR, 8%; 95% CI, 2 to 21). The Kaplan-Meier estimate of median duration of objective response was 4.5 months (95% CI, 3.7 to 4.6), and the DCR was 51% (95% CI, 35 to 68). At the data cut-off, 20 OS events were reported. The Kaplan-Meier estimate of median OS was 15.0 months (95% CI, 9.5 to not estimable).
3. The possible mechanism of AR to EGFR-TKIs
Of 39 patients who received poziotinib, mutational status in prestudy tumor tissues was determined in 37 patients (Fig. 2A), but not in two patients because of inadequate tissue samples. All tumor samples were reviewed by a thoracic pathologist (G.K.L.). Among the 37 patients, 19 (51%) had T790M mutation and two (5%) had PIK3CA mutations, and one patient had EGFR wild type.
Four patients donated tumor tissue at the end of treatment, and all were T790M-positive at screening and at the end of treatment.
To further elucidate the mechanisms of AR, plasma EGFR T790M genotyping was performed in all patients (n=39), which identified seven additional cases of T790M mutation, which were not detected in tumor tissue samples. The performance of plasma T790M genotyping is summarized in Supplementary Table 1.
Sufficient tumor tissue was available from 35 patients for MET, EGFR, and HER2 FISH analysis. Among three patients with EGFR amplifications, two overlapped with EGFR T790M mutation. No MET or HER2 amplification was detected. Of 35 patients tested using IHC, five patients had MET overexpression and four overlapped with EGFR T790M mutation. The frequencies of possible AR mechanisms are shown in Fig. 2B. Overall, of 39 patients, 26 patients (67%) developed AR to prior EGFR-TKIs due to acquisition of T790M mutation.
4. Clinical outcome according to mutational status
In subgroup analysis on the EGFR T790M mutational status in the prestudy tumor tissue, the estimated median PFS in patients with EGFR T790M mutation (2.7 months; 95% CI, 1.7 to 3.9) was shorter than that for patients without T790M mutation (3.7 months; 95% CI, 1.4 to 5.4; p=0.329) (Supplementary Fig. S1A). Similarly, patients with the EGFR T790M or PIK3CA mutations in prestudy tumor tissue had a shorter median PFS (1.9 months; 95% CI, 0.9 to 3.6) than those without these mutations (3.7 months; 95% CI, 1.7 to 5.5; p=0.103) (Supplementary Fig. S1B). However, these differences were not statistically significant.
Regarding the EGFR T790M mutation in baseline plasma samples, a significantly shorter median PFS (1.8 months; 95% CI, 1.7 to 1.9) was observed for patients with EGFR T790M mutations than for those without these mutations (3.7 months; 95% CI, 3.5 to 3.8; p=0.006) (Supplementary Fig. S1C). When combining the mutation results in tumor and plasma, a more prominent difference in PFS was observed. Patients with T790M or PIK3CA mutation (1.8 months; 95% CI, 1.7 to 1.9) had significantly shorter estimated median PFS than those without these mutations (5.5 months; 95% CI, 3.1 to 7.8; p=0.003) (Supplementary Fig. S1D).
5. Safety and tolerability
Patients remained on poziotinib for a median of 58 days (range, 17 to 443 days); 23% of patients (9/39) continued poziotinib without dose reduction throughout the treatment and others (77%) required at least one dose reduction; 15 (38%) required one dose reduction, and 15 (38%) required two dose reductions.
All patients treated with poziotinib experienced at least one treatment-related AE; the most frequently reported AEs (≥ 30% of patients) included diarrhea, rash, pruritus, stomatitis, paronychia, decreased appetite, mucosal inflammation, dry skin, and fatigue (Table 3). The most frequently reported grade 3 AEs (reported in 95% of patients) were rash, mucosal inflammation, stomatitis, decreased appetite, dermatitis acneiform, diarrhea, and hypokalemia. Overall, 17 serious adverse events were reported in eight patients (21%), with diarrhea being the most frequent (4/39; 10%) followed by stomatitis and rash (2/39; 5%, each). Rash was the most common reason for dose reduction (17/39; 44%) and dose interruption (9/39; 23%). Two events (one myositis and one rash) resulted in permanent discontinuation and there was no treatment-related death. Poziotinib was not considered to be related to any significant changes in laboratory tests, vital signs, or other safety endpoints (including chest radiography, ECG, and LVEF).
Discussion
This study assessed the efficacy and safety of poziotinib, a pan-EGFR-TKI, in patients with EGFR-mutant NSCLC who developed AR to gefitinib or erlotinib. Patients enrolled in this trial were required to meet the eligibility criteria based on Jackman’s clinical definition of AR to EGFR-TKIs in NSCLC [10]. Similar to other reports, T790M mutation was the most common cause of AR. Other mechanisms included PIK3CA mutation, EGFR amplification, or MET overexpression. In this heterogeneous patient population, overall ORR was 8% and DCR was 51%. The Kaplan-Meier estimate of median PFS (2.7 months) did not reach the extent of the original assumption used for calculation of the sample size (4.5 months). However, patients without T790M mutation had better median PFS than those with T790M mutation. These results suggest that poziotinib may not overcome AR secondary to T790M mutation. In addition, when combining the results of patients with or without T790M or PIK3CA mutations in tissue or plasma, patients without these mutations had significantly longer PFS than those with these mutations (5.5 months vs. 1.8 months, p=0.003), which exceeds the primary endpoint results of this study. This finding suggests that poziotinib may not overcome AR due to EGFR T790M or PIK3CA mutation. However, it can be considered as a new treatment option for patients who develop AR not due to these mutations.
As an irreversible EGFR-TKI, poziotinib has shown in vitro activity for EGFR T790M mutant NSCLC cells. Several irreversible EGFR-TKIs including afatinib, dacomitinib, and neratinib have been evaluated in patients previously treated with gefitinib or erlotinib, yielding a response rate of less than 10% and PFS of less than 4 months [15-17]. Despite their preclinical activity against T790M mutation, these drugs failed to demonstrate adequate efficacy in these populations. In addition, their potent wild type EGFR-inhibiting activity leads to intolerable skin and gastrointestinal toxicities, and poziotinib was associated with serious skin rash and diarrhea. Finally, most patients required frequent dose reduction, which may further decrease the potential for inhibiting T790M mutation.
Recent progress has been made in the treatment of EGFR T790M mutant NSCLC with mutant-selective EGFR-TKIs including AZD9291, rociletinib, and HM61713. Response rates of over 50% have been reported in patients with T790M mutation. In addition, compared with other EGFR-TKIs developed so far, these drugs have more favorable toxicity profiles [18-20]. Thus, these mutant-selective EGFR-TKIs would be a promising treatment option for patients with EGFR T790M mutant NSCLC.
Molecular analysis at the time of progression on EGFR-TKI therapy is important in deciding on the next treatment plan. Tumor tissue is the preferred sample type when available; however, there is increasing evidence that plasma can be used as a substitute for molecular analysis in NSCLC [21]. In this study, we also examined the utility of plasma samples for EGFR T790M mutation analysis. EGFR T790M mutation rates were higher in tumor tissue (19/37, 51% in evaluable samples) than in plasma (18/39, 46%). The concordance was 62%, with a specificity of 67% and sensitivity of 58% (Supplementary Table 1). Despite the low concordance rate observed here, the plasma T790M results were more predictive for efficacy compared with tissue results (Supplementary Fig. S1C), and five out of six patients with T790M-positive plasma but T790M-negative tissue showed PD with poziotinib. Thus, further clinical evaluation of the plasma assay is required.
Conclusion
In this explorative phase II study poziotinib provided modest clinical benefit in patients with advanced or metastatic lung adenocarcinoma having progressed on erlotinib or gefitinib. This study provided no obvious clinical evidence showing that poziotinib may overcome AR secondary to EGFR T790M mutation.
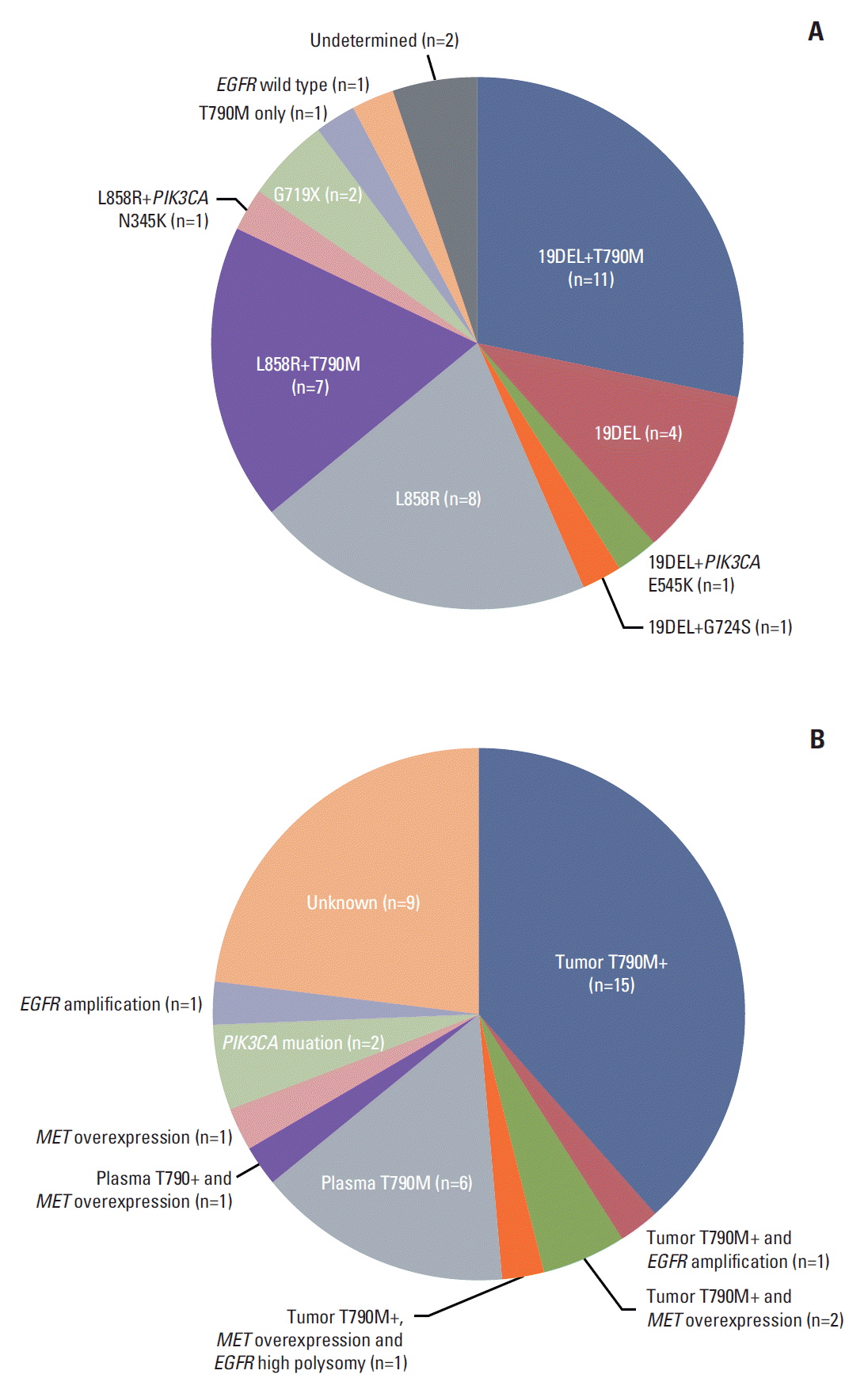
 XML Download
XML Download