

 PDF
PDF Citation
Citation Print
Print


Introduction
Esophageal carcinoma is a common malignant tumor worldwide [1,2]. Indeed, more than 300,000 people worldwide died from esophageal cancer, of which 150,000 were Chinese in 2012 [3,4]. Current treatments for esophageal cancer include surgery, chemotherapy, and radio-chemotherapy; however, there all have limited effects on the disease. Therefore, it is essential to find new curative approaches to treatment of esophageal cancer.
Several studies [5,6] have shown a close link between type 2 diabetes and the risk of some solid cancers. Patients with type 2 diabetes show increased risk of developing a range of different cancers, including colorectal, breast, and liver cancer [6-9]. Hyperinsuliemia [10] plays a crucial role in increasing cancer risk for people with type 2 diabetes. Metformin, an anti-diabetic drug, has been extensively employed for the treatment of type 2 diabetes to reduce blood glucose concentration and increase insulin sensitivity. These factors are likely to be associated with increased cancer risk. Accumulating evidence [11-13] has shown that diabetic patients treated with metformin display a reduced incidence of neoplastic disease and might improve cancer prognosis, including that of human esophageal cancer cell (ESCC), in clinical trials. Metformin [14-16] also shows evident inhibitory and pro-apoptotic effects in mice tumor models. However, the antitumor mechanisms of metformin remain elusive.
Previous reports [17,18] have shown that the antitumor mechanisms of metformin were partly attributed to activation of AMP-activated protein kinase (AMPK). AMPK can suppress the expression of mammalian target of rapamycin (mTOR), which can negatively regulate tumor growth and promote cancer cells apoptosis [19]. Furthermore, it has been argued [20] that metformin affects the insulin/insulin-like growth factor (I/IGF) pathway and decreases the expression of insulin-like growth factor 1 receptor (IGF-1R). IGF-1R [20,21], a receptor tyrosine kinase, is over-expressed in several types of cancer, including human ESCC. IGF-1R can activate the phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling pathway to regulate cell proliferation, survival, and angiogenesis. Moreover, IGF-1R can be used as an indicator and targets for cancer therapy. Pyruvate kinase muscle isozyme 2 (PKM2), which is a downstream molecule in the PI3K/AKT/mTOR pathway that plays a crucial role in the Warburg effect, is overexpressed in nearly all tumor cells. Studies [22-24] have shown that PKM2 contributes to tumor growth, which may partly be attributed to its involvement in cellular energy control and glucose metabolism. PKM2 depletion decreases the ability of human tumor cell lines to form tumors [25]. Inhibiting the PI3K/AKT/mTOR pathway may down-regulate PKM2 expression and alter cancer metabolism. Therefore, metformin could decrease the expression of IGF-1R to inhibit the PI3K/AKT/mTOR/PKM2 signaling pathway and suppress tumor growth, which may be a novel mechanism in human ESCC cancer.
In this study, we evaluated the antitumor activity of metformin in human ESCC cells. The results showed that metformin inhibited proliferation of ESCC cells, as well as induced cell cycle arrest and apoptosis. Furthermore, we found that metformin activated caspase 3, downregulated caspase 9, and increased the pro-apoptotic protein, Bim. To explore the underlying mechanisms, we observed the role of the PI3K/AKT/mTOR pathway in metformin mediatedantitumor response and found that it decreased the expression of IGF-IR, PI3K, AKT, pAKT, mTOR, and PKM2. IGF-1 partly reversed the antitumor effect of metformin in vitro. To explain determine if the down-regulation of PKM2 is associated with up-regulation of Bim, we depleted the PKM2 gene in ESCCs. We found that knockout PKM2 resulted in activation of caspase 3, downregulation of caspase 9 and increased expression of Bim. In addition, metformin reduced tumor burden in an ESCC xenograft animal model. In conclusion, our results supported that metformin has the ability to inhibit the proliferation of ESCCs, which may lead to suppression of the PI3K/AKT/mTOR signaling pathway.
Go to : 
Materials and Methods
1. Cell culture and regents
Eca109 and EC9706 were provided from American Type Culture Collection. Cells were maintained in 1640 media containing 10% fetal bovine serum in a humidified incubator at 37°C under 5% CO2.
Metformin was purchased from Sigma (San Francisco, CA). Antibodies against caspase 3, cleaved caspase 3, caspase 9, Bcl-2, Bid, Bim, and PKM2 were purchased from Abcam (Cambridge, MA). IGF-IR, PI3K, AKT, pAKT, mTOR, and p70S6K were purchased from Cell Signaling Technology (Danvers, MA). β-Actin and rabbit secondary antibody were purchased from ZSGB-Bio (Beijing, China). A TUNEL staining In Situ Cell Death Detection Kit was obtained from Promega (Madison, WI). Cell Counting Kit-8 (CCK-8) was purchased from Nan Jing Jian-Cheng Bioengineering (Nanjing, China).
2. CCK-8 viability assay
The inhibitory effect of metformin was assessed by cell viability using CCK-8 assay. Cells were grown in 96-well plates at 5×103 cells per well and cultured overnight at 37°C. Different concentrations of metformin were used to treat cells for 24 hours and 48 hours. The absorbance was determined at 450 nm using microplate reader.
3. Flow cytometric assay
Cells were collected and seeded into 6-well plates at 4×105 cells per well, then maintained for 24 hours. Next, cells were treated with metformin (10 and 20 mM) for 24 hours. Cell cycle and apoptosis were determined by flow cytometry according to the manufacturer’s protocol (Boster, Wuhan, China). The results were indicated as the mean±standard diviation.
4. Western blot
Cells were collected and lysed using RIPA buffer (ZSGB-Bio) containing phenylmethanesulfonylfluoride. Total protein was extracted and determined using a BCA Protein Assay Kit. Total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, then transferred to polyvinylidene fluoride membrane. The first antibodies were added at the proper dilution and incubated at 4°C overnight, after which the appropriate horseradish peroxidase (HRP)–conjugated secondary antibody was added at the appropriate dilution and incubated for 60 minutes. Finally, the bands were examined by enhanced chemiluminesence reagents.
5. Transfection with small interference RNA
The PKM2 small interference RNA (siRNA) and control were obtained from Genepharma (Shanghai, China). Eca109 or EC9706 cells were seeded in a 12-well plate (3×105 cells/well) and transfected with 50 nM siRNA using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA). At 48 hours after transfection, the total cell extracts were collected for western blot, which was conducted using the siRNAs listed as follows: PKM2: siRNA 5′-GGCUUCUUAUAAGGUUUATT-3′; negative control 5′-UUCUCCGAACGUGUCACGUTT-3′.
6. Real-time fluorescent quantitative polymerase chain reaction
Trizol reagent (Bioo Scientific Co., Austin, TX) was used to extract the total RNA from cultured cells (Eca109 and EC9706). β-Actin and Bim mRNA were first reversed to generate a first-strand synthesis of DNA. Real-time fluorescent quantitative polymerase chain reaction (PCR) was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) with the following primers: Bim: forward 5′-TAAGTTCTGAGTGTGACCGA-GA-3′, reverse 5′-GCTCTGTCTGTAGGGAGGTAGG-3′; β-actin: forward 5′-ATGCCAACACAGTGTTGTCTGG-3′, reverse 5′-TACTCCTGCTTGCTG ATCCACAT-3′.
7. Tumor xenograft model
Nude mice (6-week old) were used to establish an Eca109 tumor model. First, Eca109 cells were harvested, after which the mice were injected subcutaneously with 5×106 tumor cells in the dorsal area. The mice were then randomly divided into control and treatment groups (5 mice per group) after 7 days. Next, mice in the treatment group were given metformin (300 mg/kg) via intraperitoneal injection, while those in the control group were administered normal saline (100 μL). Treatments were performed for 21 days, during which time tumor volumes (v=0.52×width2 ×length) were determined every 3 days.
8. Immunohistochemistry
Tumor tissues were embedded in paraffin, then sectioned (4 μm). Next, sections were deparaffinized, rehydrated with phosphate buffered saline containing H2O2 (3%). Immunostaining was then undertaken using anti-mouse PKM2 (1:100) and Bim (1:100) monoclonal antibody at 4°C. HRP-labeled second antibody was then used to combine with the primary antibody. An HRP-IgG antibody kit (Boster) was employed according to the manufacturer’s instructions and immunostaining results were quantified by counting cells positively stained with PKM2 and Bim in six consecutive and independent fields near the center of each section.
9. Quantitative assessment of apoptosis
Tumor tissues were fixed in 10% formalin, then embedded in paraffin, after which apoptosis was assessed using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; DeadEnd Fluorometric TUNEL System, Promega) according to the manufacturer’s instructions.
10. Statistical analysis
Data were analyzed by t tests conducted using SPSS ver. 17.0 (SPSS Inc., Chicago, IL). A p < 0.05 was considered to indicate significance.
Go to :
Results
1. Metformin inhibited the proliferation of ESCCs
Eca109 and EC9706 cells were treated with different concentrations of metformin (0, 5, 10, 20, and 40 mM) for 24 hours and 48 hours. CCK-8 assay was used to determine the cell viability. The results showed that metformin inhibited the proliferation of ESCCs in a time and dose-dependent manner (Fig. 1A and B).
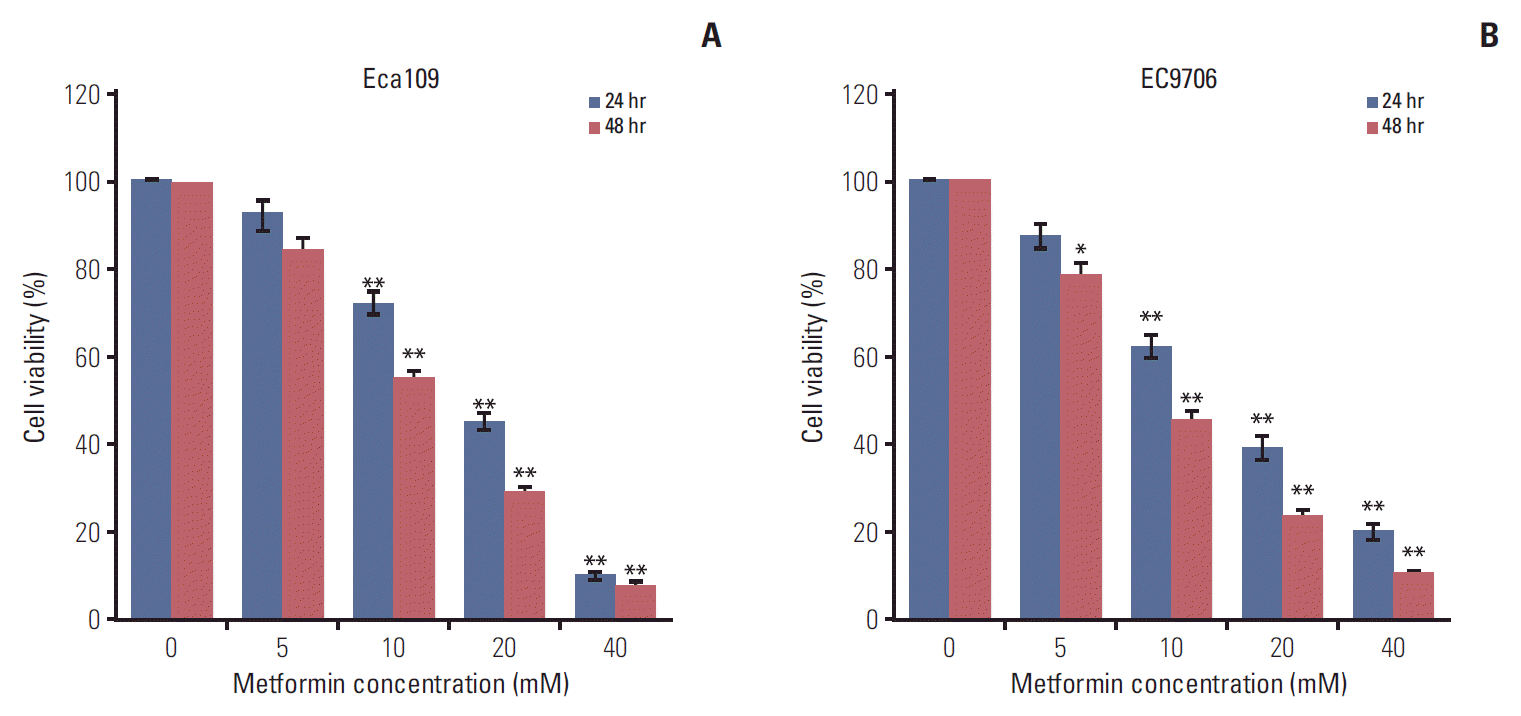 | Fig. 1.Metformin inhibited the proliferation of esophageal cancer cells. Eca109 and EC9706 cells were treated with metformin (0, 5, 10, 20, and 40 mM) for 24 hours and 48 hours. Cell viability was determined by Cell Counting Kit-8. (A) Metformin reduced Eca109 cell viability. (B) Metformin decreased EC9706 cell viability. Data represent the mean±standard deviation of three independent experiments (*p < 0.05, **p < 0.01).
|
2. Metformin induced G0/G1 phase cell cycle arrest and apoptosis
To explore the possible mechanisms through which metformin suppressed the growth of ESCC, cell cycle and apoptosis were determined by flow cytometry after metformin treatment. When compared to the control, G0/G1 cells had accumulated (Fig. 2A and B) and apoptosis cells had increased in the experimental group (Fig. 2C and D).
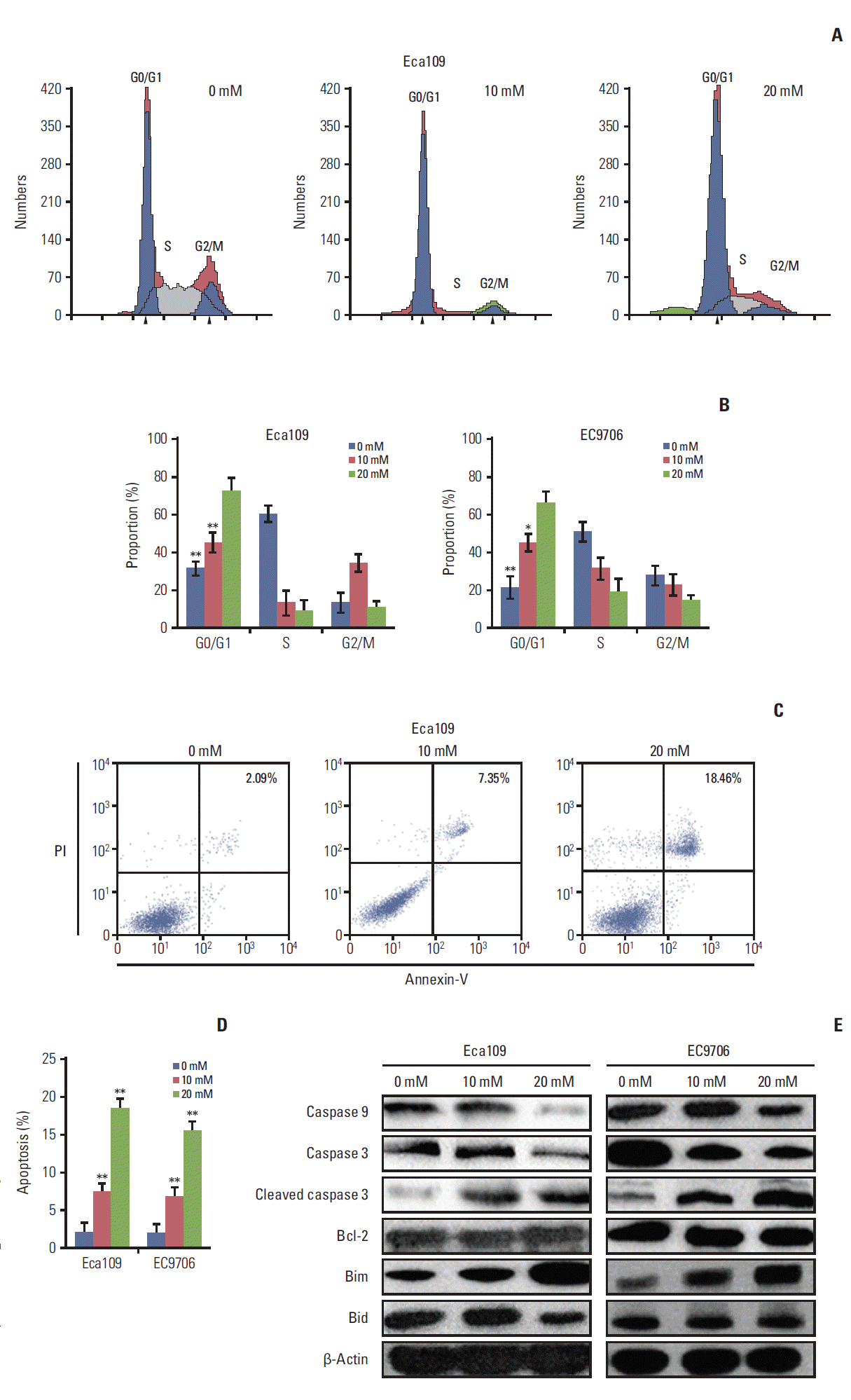 | Fig. 2.Metformin induced cell cycle arrest and apoptosis in esophageal cancer cell cells. Eca109 and EC9706 Cells were treated with metformin (10 and 20 mM) for 24 hours. To investigate the cell cycle, cells were stained with propidium iodide (PI) according to the manufacturer’s instructions. For apoptosis determination, cells were analyzed by flow cytometry after staining with Annexin-V/PI according to the manufacturer’s instructions. (A) Typical flow cytometric graph of cell cycle. (B) The percentage of cells in each cell cycle phase. The results showed that metformin blocked cell arrest in the G0/G1 phrase in Eca109 and EC9706. (C) Typical flow cytometric graph of apoptosis. (D) The percentage of apoptotic cells. The results showed that metformin increased the rate of apoptosis in Eca109 and EC9706. (E) The expression of apoptosis-related proteins. Eca109 and EC9706 were treated with metformin. At 24 hours after treatment, the cells were lysed and subjected to immunoblot analysis. The levels of caspase 3, caspase 9, cleaved caspase 3, Bcl-2, Bim, and Bid were detected. Data represent the mean±standard deviation from three independent experiments (*p < 0.05 and **p < 0.01 compared to control, n=3).
|
The apoptosis-related proteins expression were detected by western blot. The results clearly showed that metformin activated caspase 3, down-regulated caspase 9, and up-regulated Bim. Bcl-2 and Bid remained unchanged (Fig. 2E).
3. Metformin suppressed PI3K/AKT/mTOR signaling pathway
To examine the potential molecular mechanisms for metformin antitumor effects, we detected the expression of IGF-IR, PI3K, AKT, pAKT, mTOR, p70S6K, and PKM2 by western blot. The results revealed that metformin reduced IGF-IR, PI3K, AKT, pAKT, mTOR, p70S6K, and PKM2 in EC109 and EC9706 cells (Fig. 3A and B).
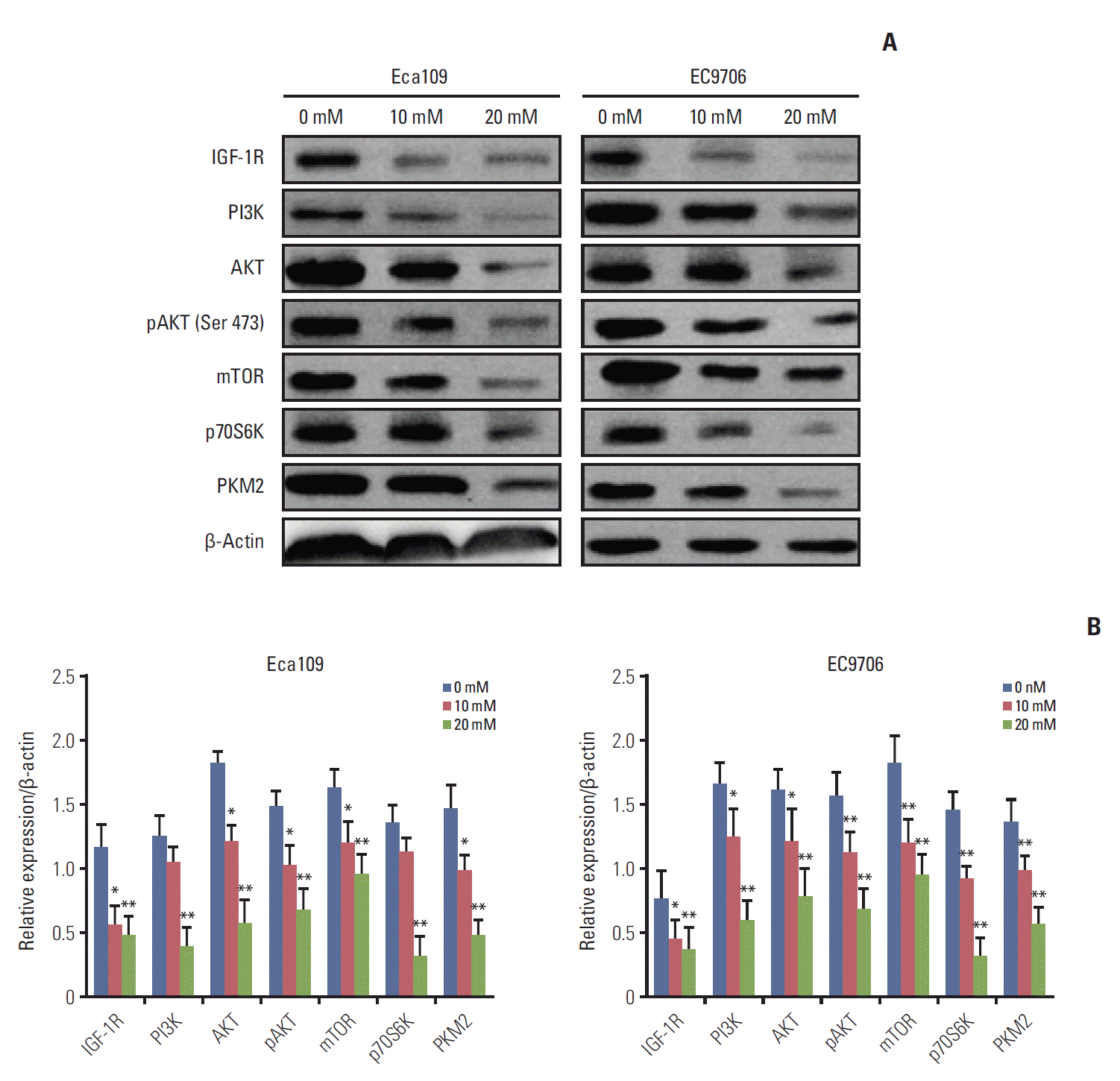 | Fig. 3.Metformin suppressed the PI3K/AKT/mTOR signaling pathway. Eca109 and EC9706 cells were treated with metformin (0, 10, and 20 mM) for 24 hours. Total proteins were extracted as previously described. (A) Western blot analyses of IGF-1R, PI3K, AKT, pAKT, mTOR, p70S6K, and PKM2. Metformin reduced the IGF-1R expression and suppressed the PI3K/AKT/mTOR/PKM2 pathway. (B) Graph depicts densitometric analyses of IGF-1R, PI3K, AKT, pAKT, mTOR, p70S6K, and PKM2 normalized by β-actin (*p < 0.05 and **p < 0.01 compared to control, n=3). PI3K, phosphoinositide 3-kinase; pAKT, phosphorylation of AKT; mTOR, mammalian target of rapamycin; PKM2, pyruvate kinase muscle isozyme 2; IGF-1R, insulin-like growth factor 1 receptor.
|
4. IGF partly reversed the effect of metformin-induced apoptosis and attenuated the repression effect of metformin on the PI3K/AKT/mTOR/PKM2 pathway
To determine whether the effects of metformin mediated-apoptosis on Eca109 and EC9706 cells were associated with the IGF-IR/PI3K/AKT/mTOR pathway, insulin growth factor (IGF) treatment (100 ng/mL) was carried out 2 hours before metformin was added, after which apoptosis was detected by flow cytometry. The results showed that IGF partly reversed the effects of metformin-induced apoptosis in Eca109 and EC9706 (Fig. 4A and B). Furthermore, IGF attenuated the repression effect of metfomin on PI3K, pAKT, mTOR, and PKM2 (Fig. 4C). These results suggest that the effects of metformin-induced apoptosis may be connected with the PI3K/AKT/mTOR/PKM2 signaling pathway.
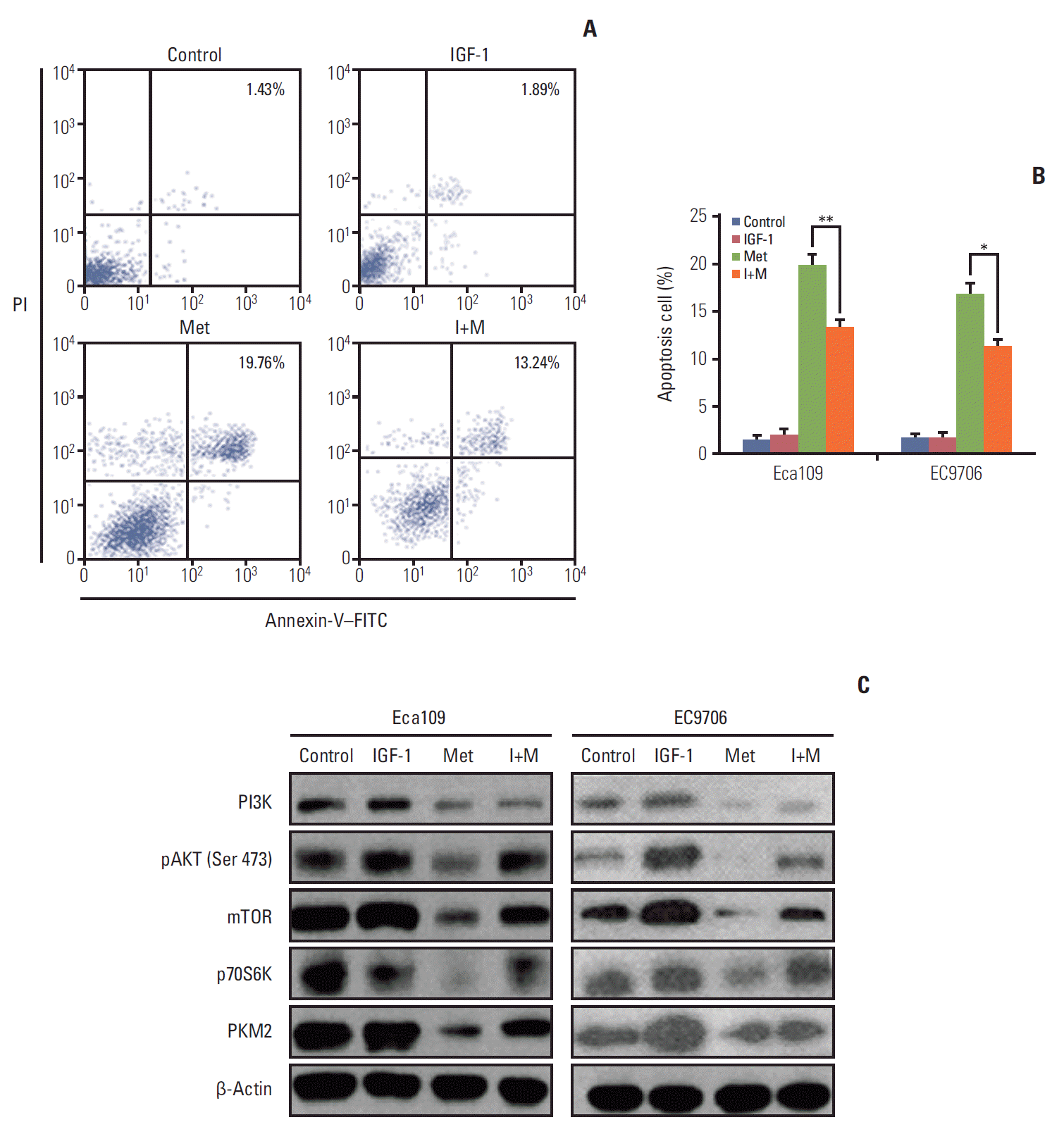 | Fig. 4.IGF partly reversed the effect of metformin-induced apoptosis and attenuated the repression action of metfomin toward PI3K, pAKT, mTOR, p70S6K, and PKM2 in esophageal cancer cells. Eca109 and EC9706 cells were pre-treated with IGF-1 (100 ng/mL) for 2 hours, then with metformin for 24 hours. Apoptosis was determined by flow cytometry. (A) Typical flow cytometric graph of apoptosis. (B) The average percentage of apoptosis in Eca109 and EC9706 cells after treatment with metformin. The results showed that metformin partly prevented the metformin-induced apoptosis. (C) The expression of PI3K, pAKT, mTOR, p70S6K, and PKM2 was detected by western blot. The outcome indicates that IGF-1 attenuates the repression action of metformin toward PI3K, pAKT, mTOR, p70S6K, and PKM2 in Eca109 and EC9706 cells (*p < 0.05 and **p < 0.01 compared to control, n=3). PI3K, phosphoinositide 3-kinase; pAKT, phosphorylation of AKT; PKM2, pyruvate kinase muscle isozyme 2; IGF-1, insulin-like growth factor 1; mTOR, mammalian target of rapamycin; PI, propidium iodide; Met, metformin; I+M, insulin-like growth factors+metformin; FITC, fluorescein isothiocyanate.
|
5. Knockdown PKM2 altered apoptosis-related protein expression
To explain the relationship between the PI3K/AKT/mTOR/PKM2 signaling pathway and apoptosis, we depleted PKM2 to detect the expression of apoptosis-related proteins in Eca109 and EC9706. Knockdown of PKM2 activated caspase 3 and down-regulated the expression of caspase 9. Bim was notably upregulated and the Bim mRNA level was increased by PKM2-specific siRNA (Fig. 5). These results were consistent with the change in apoptosis-related proteins caused by metformin. Based on our findings, metformin increased the expression of the pro-apoptosis protein Bim, partly due to the suppression of the PI3K/AKT/mTOR/PKM2 signaling pathway.
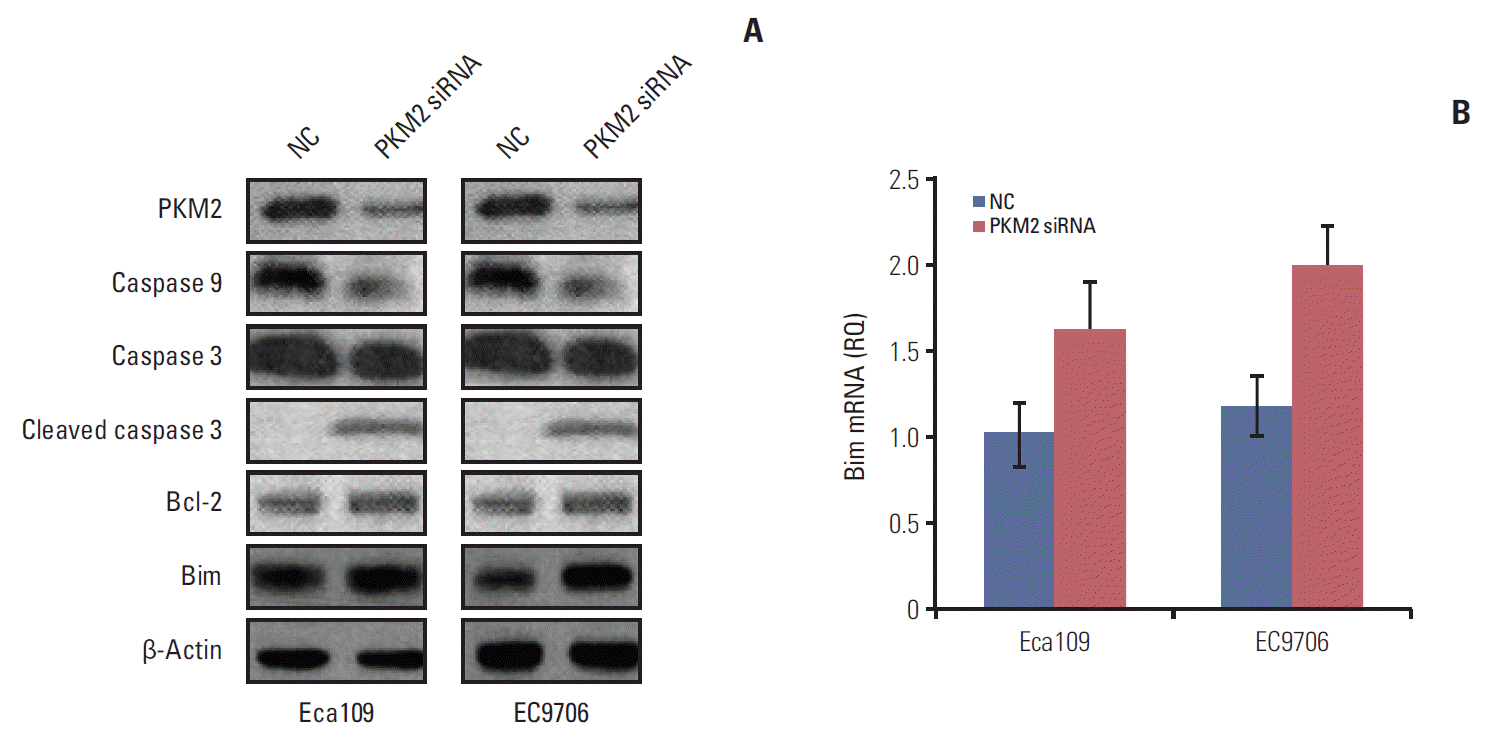 | Fig. 5.Knockdown of PKM2 altered the expression of apoptosis-related proteins. Eca109 or EC9706 cells (3×105 cells/well) were plated in a 12-well plate. PKM2 was knocked down in Eca109 and EC9706 cells by siRNA (50 nM final concentration for both PKM2-specific and NC siRNA). At 48 hours after transfection, the whole cell extracts were collected for western blot. Total RNA was extracted from cultured cells (Eca109 and EC9706) using Trizol reagent according to the manufacturer’s instructions. β-Actin and Bim mRNA were quantified in duplicate by SYBR Green 2-step, real-time reverse transcription polymerase chain reaction. (A) Caspase 9, caspase 3, cleaved caspase 3, Bcl-2, and Bim were detected by western blot. The results showed that knockout PKM2 activated caspase 3 and downregulated caspase 9. Bim protein expression was evidently increased. (B) Bim mRNA level was increased in depletion PKM2 esophageal cancer cells. PKM2, pyruvate kinase muscle isozyme 2; NC, normal control.
|
6. Antitumor effect of metformin in vivo
To further assess the effects of metformin on ESCC proliferation in vivo, an Eca109 tumor model was established in nude mice. We found that metformin treatment decreased the tumor burden and that the inhibition rate reached up to 48.41% in the experimental group when compared with the control (Fig. 6A and B). Furthermore, we found that metformin reduced the expression of PKM2 and increased the expression of Bim in the tumor tissue (Fig. 6C and D). Moreover, evaluation of apoptosis by TUNEL staining revealed that there were more apoptotic cells in the experimental group than the control (Fig. 6E and F). Overall, these results indicate that increasing apoptosis may be related to the antitumor effect of metformin in the Eca109 model.
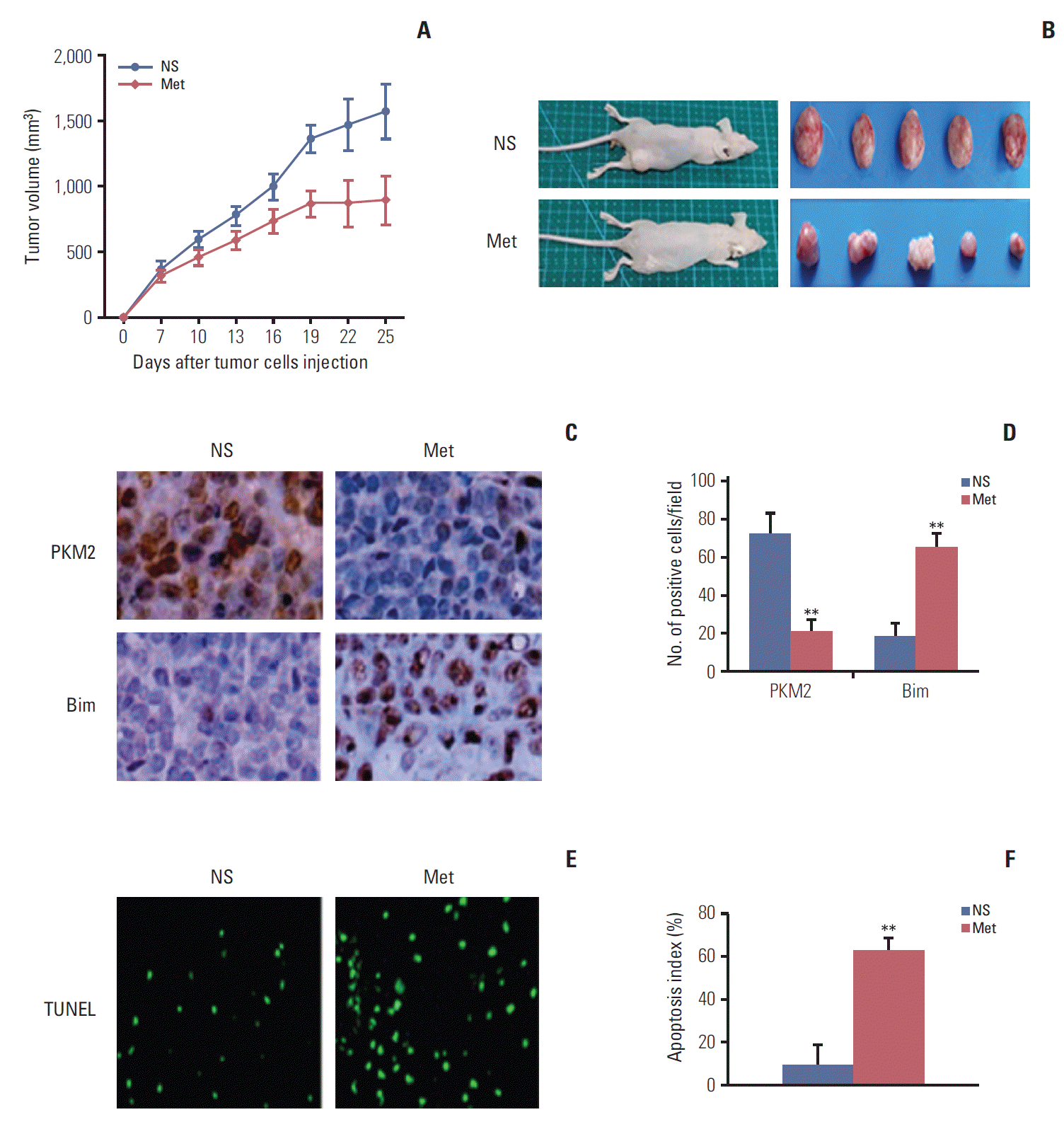 | Fig. 6.Metformin inhibited tumor growth in vivo. Mice were treated with NS or metformin (300 mg/kg/day) for 21 days at the same time (n=5 per group). (A) Suppressed growth of tumor in mice. The results showed that metformin inhibited tumor growth (n=5, p < 0.01). Data are shown as the mean±standard deviation. (B) Representation of photos of tumors from mice bearing Eca109 receiving different treatments. Mice were sacrificed when they were moribund. (C) Typical graph of immunohistochemical staining of PKM2 and Bim in tumor tissues (×400). (D) Quantification of the number of PKM2 and Bim positive cells per field. Metformin treatment significantly decreased the number of PKM2-positive cells and increased the number of Bim-positive cells. (E) Typical sections from tumor tissue. (F) Apoptotic index within tissues. Metformin showed a significant increase of apoptotic cells in tumor tissues compared to the control (**p < 0.001). Data are expressed as the mean apoptotic index±standard deviation of cancer cells. NS, normal saline; PKM2, pyruvate kinase muscle isozyme 2; Met, metformin; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
|
Go to :
Discussion
In this study, we investigated the antitumor effects of metformin on ESCCs in vitro and in vivo. The results showed that metformin decreased the cell viability (Fig. 1) and induced cell cycle arrest and apoptosis (Fig. 2). Furthermore, metformin activated caspase 3, decreased caspase 9, and increased the expression of Bim. Potential mechanisms of antitumor activity have been investigated, and the results have shown that they may be associated with inhibition of the PI3K/AKT/mTOR/PKM2 signaling pathway (Fig. 3). IGF partly reversed the effects of metformin-mediated apoptosis, further confirming that metformin exerts its anti-tumor effects in connection with the PI3K/AKT/mTOR/PKM2 signaling pathway (Fig. 4). To explore the association between suppressing PI3K/AKT/mTOR/PKM2 and apoptosis, apoptosis-related proteins were detected after knockdown of PKM2 in Eca109 and EC9706 cells. We found that PKM2 depletion activated caspase 3 and reduced caspase 9. Bim protein expression was substantially increased and Bim mRNA level was up-regulated after PKM2 depletion (Fig. 5). In vivo, we found that metformin reduced tumor burden (Fig. 6A and B), and that this was accompanied with down-regulation of PKM2 and up-regulation of Bim in the tumor tissue (Fig. 6C and D). To the best of our knowledge, this is the first study to demonstrate that the PI3K/AKT/mTOR/PKM2 signaling pathway is a target of metformin in human ESCCs.
Although previous studies [26,27] showed that metformin inhibited proliferation of ESCC, the mechanisms responsible for this effect remained controversial. Currently, there are two main theories regarding that the mechanism through which metformin exerts its effects. In one, its effects are believed to occur through AMPK dependent and AMPK independent mechanisms under different cellular settings. It has been suggested [26,27] that metformin affects the AMP/ATP balance and active AMPK, which could negatively regulate mTOR signaling, further inhibiting protein synthesis and regulating the phosphorylation of different proteins. The other possibility [28,29] is that antitumor effects are linked to the ability of metformin to inhibit transcription of key gluconeogenesis genes in the liver and stimulate glucose uptake in muscle, increasing insulin sensitivity and reducing blood glucose while lowering the insulin level. A recent report [30] showed that metformin promoted autophagy and apoptosis in ESCC by down-regulating the STAT3 signaling pathway.
In the present study, we corroborate and extend the results of previous studies. We mainly investigated whether metformin affects the I/IGF pathway to inhibit the proliferation of ESCCs. Overexpression of IGF-1R [20,31] has been reported in human esophageal carcinoma cells. Specifically, overexpression can be activated by IGF-1 or IGF-2, which induces the PI3K/AKT/mTOR signaling pathway, resulting in survival and proliferation in various cancer cells. Thus, inhibition of IGF-1R [21] may be effectively block tumor growth in human ESCC. Our results verify that metformin down-regulates the expression of IGF-1R, which suppresses the PI3K/AKT/mTOR/PKM2 pathway in ESCCs. IGF can partly block metformin-induced anti-tumor effects and attenuate the repression action of metfomin toward PI3K, pAKT, mTOR, p70S6K, and PKM2. PKM2, a downstream molecule of PI3K/Akt/mTOR signaling pathway, plays a crucial role in regulation of the metabolic fate of glycolytic intermediates for cancer cells [32]. Knockdown of PKM2 [33] caused increased apoptosis in many cancer cell lines. Inhibition of PI3K/AKT/mTOR down-regulates PKM2 expression and suppresses cancer metabolism, which has been confirmed in other various cancer cell lines. We found that metformin decreased the expression of PKM2 in esophageal cancer, suggesting that reducing PKM2 expression represents a general mechanism of metformin cytotoxicity, not limited for gastric cancer [34] and breast cancer [35]. In order to explain the association between PKM2 and apoptosis, we found knockdown PKM2 activated caspase 3 and down-regulated caspase 9. The pro-apoptosis protein Bim increased after PKM2 depletion. Overall, our results revealed that induction of apoptosis by metformin may be associated with down-regulation of PKM2 in human ESCC cells.
We further confirmed that metformin decreased the tumor burden in vivo, showing that metformin treatment may be effective against ESCC. Following metformin treatment, down-regulation PKM2 and up-regulation Bim were consistent with the previous in vitro results. We also observed that metformin induced tumor cells apoptosis. Most importantly, there were no significant adverse effects in the metfromin-treated mice. These findings indicate that metformin has potential antitumor effects in human ESCC.
However, it should be noted that the present study has some limitations. Specifically, we are not sure how metformin affects the expression of PKM2. It may bind PKM2 directly or indirectly influence the expression of PKM2; therefore, further study is needed. Moreover, the high dose of metformin used in this experimental setting may not be easy to transform to a clinical application. Accordingly, more clinical trials are needed to confirm the anti-tumor effects of metformin.
Go to :
Conclusion
In conclusion, our results confirm that metformin inhibits the proliferation of ESCCs in vitro and vivo. These effects may be attributed to metformin inducing cell cycle arrest and apoptosis. The molecular mechanisms are connected with inhibition of the PI3K/AKT/mTOR/PKM2 signaling pathway. Our results support that metformin may become a novel and effective therapeutic agent for the treatment of esophageal cancer.
Go to :
 XML Download
XML Download