

 PDF
PDF Citation
Citation Print
Print


Introduction
von Hippel-Lindau disease (VHL) is a hereditary, autosomal-dominant, neoplastic disease associated with various tumor types, including central nervous system (CNS) and retinal hemangioblastomas, clear cell renal carcinomas, pheochromocytomas, and pancreatic neuroendocrine tumors, in addition to pancreatic and renal cysts. The disease is caused by mutations in the VHL tumor-suppressor gene. As described by the two-hit hypothesis of Knudson, affected patients inherit one mutated copy of the VHL gene from an affected parent through the germline. Later in life, the other normal copy of the VHL gene undergoes somatic mutation in susceptible tissues (i.e., the second hit), initiating local tumorigenesis. The VHL protein is expressed ubiquitously, and its function is primarily associated with the formation of a ubiquitin ligase complex with other participating proteins (i.e., elongin B and elongin C) that subsequently binds and directs the degradation of the transcription factor hypoxiainducible factor (HIF). Loss of VHL function in mutated cells is mainly caused by dysregulated accumulation of HIF, which then directs the excessive transcription of downstream genes, including angiogenic growth factors such as vascular endothelial growth factor and platelet-derived growth factor [1].
Carnitine palmitoyltransferase type II (CPT2) deficiency is an autosomal recessive disorder representing the most common inherited disorder of long-chain fatty acid oxidation affecting skeletal muscle. During prolonged exercise, fasting, exposure to cold, fever, emotional stress, and drugs, production of extra energy demand is met by oxidation of fatty acids. Under these conditions, long-chain fatty acids are the main source of energy substrate of muscle. However, longchain fatty acids do not readily diffuse across the mitochondrial membrane and hence require trans-esterification to acylcarnitine. Formation of acylcarnitine from carnitine and long-chain fatty acyl-CoA is catalyzed by CPT1 at the outer mitochondrial membrane and then crosses the inner mitochondrial membrane. At the inner side of the inner mitochondrial membrane, CPT2 catalyzes formation of acyl-CoA, which is then available for β-oxidation. There are three phenotypes of CPT2 deficiency: lethal neonatal form, severe infantile hepatocardiomuscular form, and mild myopathic form [2]. In the most frequent muscle form, recurrent attacks of myalgias and cramps, often associated with myoglobinuria are the clinical hallmark. Rhabdomyolytic episodes, usually induced by the above-mentioned triggering factors, may be complicated by life-threatening events, including acute renal failure (ARF; not specific of CPT2 deficiency, but consequent to myoglobinuria of any cause), respiratory failure, and, more rarely, cardiac arrhythmias, and hypoglycemia [3].
Herein, we describe, for the first time, the coexistence of VHL disease and CPT2 deficiency and its consequences on the clinical manifestation.
Case Report
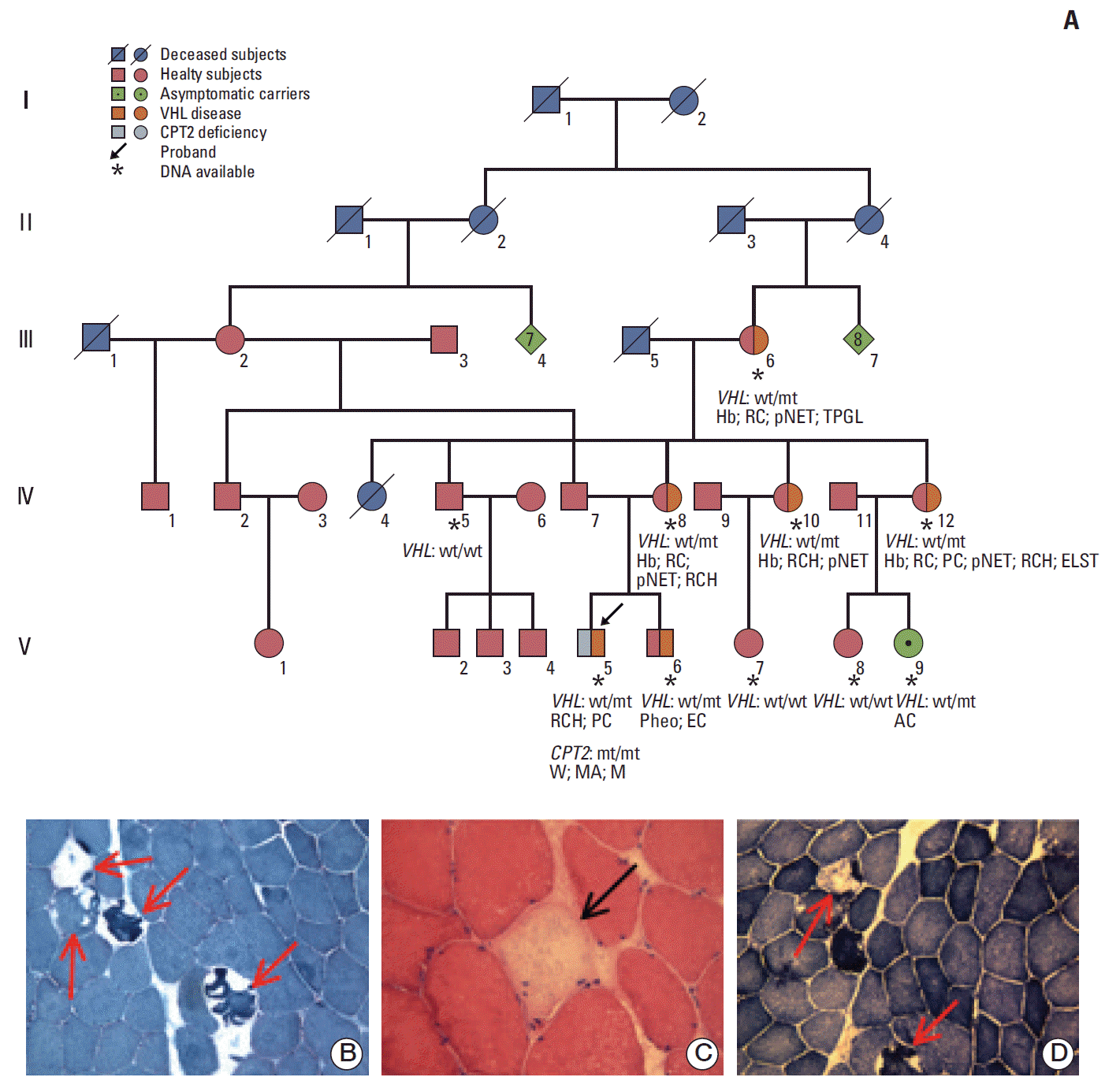
A 25-year-old male patient (V-5) (Fig. 1A), former, non-professional, soccer player was born to consanguineous parents. At the age of 10 years, familial history of VHL disease prompted mutational analysis of the VHL gene. He was found to be a VHL carrier (chromosome 3p25.3; c. 256 C>G; p.P86A mutation) without VHL-related manifestations at the time of diagnosis but later developing bilateral retinal capillary hemangioblastomas and multiple pancreatic simple cysts. The patient was hospitalized in 2003, 2006, and 2008 at ages of 12, 15, and 17 years, respectively, for diffuse muscular pain, muscle weakness, and brown-colored urine. Symptoms in all three episodes were triggered by prolonged exercise in addition to acute febrile illness caused by pneumonia (in 2003 and 2006) and gastroenteritis (in 2008) and resolved within a few days with antibiotics therapy. At each hospitalization, neurological examination was normal whereas serum creatine kinase, aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and serum and urine myoglobin levels were abnormal (Table 1). On the contrary, serum creatinine and potassium were completely normal during (Table 1) and between the attacks. The recurrent attacks suggested an inherited cause of rhabdomyolysis. Electromyography showed no abnormality whereas quadriceps muscle biopsy showed necrosis of muscle fibers (“ghost fibers”) (Fig. 1B-D). With suspicion of a CPT2 deficiency, biochemical CPT2 assay showed a marked CPT deficiency (isotope, 43.8 pmol/min/mg [normal value, 452±160 pmol/min/mg]; forward, 137 pmol/min/mg [normal value, 367±110 pmol/min/mg]) along with elevation of serum long-chain acylcarnitines levels (Table 1). CPT2 gene sequencing showed the homozygous substitution c.338C>T (p.S113L). Frequent meals with carbohydrate-rich intake before exercise and restriction of long-chain fatty acid intake along with medium chain fatty acid supplementation were recommended to prevent further attacks.
Family history was negative regarding manifestations of CPT2 deficiency, whereas mother, brother, maternal aunts, and maternal grandmother of the proband presented VHL manifestations (Fig. 1A).
Discussion
This study describes the first patient with coexistence of VHL disease and CPT2 deficiency due to mutations in the VHL and CPT2 genes, respectively.
The mutation p.P86A in the VHL gene, found in the proband, his brother, mother, maternal grandmother, maternal aunts, and cousin was previously reported [4-6] and is associated with pheochromocytoma, peripherally located retinal capillary hemangioblastomas, and, in our experience, CNS hemangioblastomas and pancreatic neuroendocrine tumors. The mutation involves a hydrophobic aminoacid of the VHL protein that is essential for the structural integrity of the β domain, which binds HIF [7].
p.S113L mutation in the CPT2 gene, found in a homozygous state in the proband (V-5) (Fig. 1A), is present in approximately 70% of patients with the myopathic form of CPT2 deficiency. Most patients with this genotype present a relatively severe phenotype, often associated with life-threatening events and, occasionally, with a fatal outcome. Among patients who are compound heterozygotes for the p.S113L and one deleterious other mutation, some patients had the typical phenotype, but others experienced serious complications, further supporting that p.S113L is a potentially dangerous mutation [3].
Rhabdomyolysis is a leading cause of acute kidney injury (AKI), which is responsible for ARF [8]. Tubular ischemia is believed to be centrally involved in the initiation and establishment of AKI because intrarenal oxygen tensions are low and further reduced by perfusion [9]. Interestingly, despite a severe reduction of CPT activity (10% of control), the proband presented only severe weakness and myalgias but never manifested ARF during the rhabdomyolysis attacks. Several explanations can be offered. First, the degree of rhabdomyolysis might not be sufficient to cause AKI, even though patients with creatine kinase levels > 5,000 IU/L are at risk of developing AKI [10]. Second, the homozygous p.S113L mutation manifests with various degrees of clinical severity and large intra-familiar heterogeneity of the phenotype (ranging from asymptomatic to fatal) depending on different degree of exposure to triggering factors, in combination with other genetic factors and on the influence of intragenic polymorphisms [3]. This explains why 70% of cases carrying the homozygous p.S113L mutation have not experienced any episodes of ARF [2]. Third, and more intriguingly, the absence of AKI might be due to the protective effect of the VHL mutation against acute tubular injury. Indeed, a recent study reported that selective activation of HIF in renal tubules, through an inducible knockout of VHL protein (VHLKO), protects from rhabdomyolysis-induced AKI. In this model, HIF activation showed inverse correlation with tubular injury and was associated with activated glycolysis, cellular glucose uptake and utilization, autophagy, vasodilation, and proton removal showing that a metabolic shift toward anaerobic ATP generation is the central protective mechanism against AKI [11]. Conscious of extending findings from a VHLKO animal model to a VHL-heterozygous human condition, it is possible that in our patient the haploinsufficiency of VHL protein in renal tissue increased HIF levels to an extent similar to that in mouse thus protecting kidney from acute injury.
In conclusion, we have reported on the first case of coexistence of VHL disease and CPT2 deficiency. This case could be the first example of how a VHL mutation, in addition to other genetic and environmental factors, protects the patient from rhabdomyolysis-induced AKI.
 XML Download
XML Download