

 PDF
PDF Citation
Citation Print
Print


Introduction
Tyrosine kinase activity has been reported as a major cause of various anomalies in humans. In addition, the prototypical oncogenes usually play a key role in several types of human malignancies [1]. Chronic myeloid leukemia (CML), a clonal myeloproliferative disorder of transformed hematopoietic progenitor cells, is a consequent of specific cytogenetic abnormality in the Philadelphia chromosome owing to reciprocal translocation between the parts of two chromosomes (9th and 22nd) resulting in abnormal fusion of genes with different chromosomes. The abnormal fusion encodes a protein, p210BCR-ABL, with dysregulated tyrosine kinase functions, which induces constitutively active tyrosine activity that is mainly responsible for the pathogenesis of certain tumors and multi-proliferative disorders [2,3].
For all reported cases of adult leukemia, CML only accounts for 15% with an occurrence rate of one to two patients per hundred thousand annually [4]. CML can affect any age group, but is predominant in elderly [5]. In the majority of cases (~85%), patients are in the chronic phase of CML at the time of diagnosis [4]. The survival rate of CML patients has shown noticeable improvement owing to earlier diagnosis and treatment with allogeneic bone marrow transplantation or interferon-α (IFN-α). However, these treatment modalities have their own limitations, transplantation is allied with high mortality and post-transplantation morbidity whereas IFN-α therapy resulted in poor cytogenetic remission of 6%-20% [6] and therapy induced side effects caused forced discontinuation of treatment [7].
Constant progress in cell biology and knowledge of pathogenesis of CML led to the development of target oriented therapy. Thus, tyrosine kinase inhibition has arisen as a promising strategy that effectively modulates the specific molecular events responsible for Bcr-Abl positive leukemia. Imatinib, a phenylaminopyrimidine derivative, competitively inhibits the Bcr-Abl tyrosine kinase by blocking the interaction between adenosine triphosphate and tyrosine kinase thereby suppressing phosphorylation and activating downstream proteins. It is reported to be successful in achieving hematological responses in patients with chronic phase CML. In addition, imatinib inhibits tyrosine kinase activity of the mutated platelet-derived growth factor receptor [8] and stimulates natural killer cells [9]. Suggested mechanisms are responsible for its activity against gastrointestinal stromal tumors (GIST) which is deprived of mutated c-kit [9,10]. A pilot study conducted on 58 patients who received oral dosing of imatinib mesylate (300-1,000 mg/day) showed a response in 55% of patients of myeloid blast crisis (21 out of 38) with a 19% complete hematologic response, while lymphoid blast crisis cases showed a 70% response rate (14 out of 20) with 20% complete hematologic responses [11]. Thirty-three percent of responding CML patients were continued on imatinib treatment, which showed prolonged remission ranging from 101 to 349 days from the commencement of therapy. Based on clinical study data, Food and Drug Administration (FDA) approved Imatinib mesylate (Gleevec Novartis Pharmaceuticals, Basel, Switzerland) for treatment of patients with CML in May 2001.
Imatinib shows rapid and almost complete absorption (98%) after oral ingestion irrespective of dose (100 or 400 mg) and dosage forms like solution, capsule, or tablet [12-14] and attains its peak plasma concentration (Cmax) within 2-4 hours [15]. Its absorption is usually not affected by food, but marginally affected by fatty diets. Fatty diets result in a minor reduction in peak plasma concentration (11%), and extent of absorption (7.4%) with a delay of up to 1.5 hours to attain peak plasma concentration (Tmax) [13,16]. Imatinib exhibits cytochrome P4503A4 (CYP3A4) mediated metabolism and generates a major metabolite, N-demethylated piperazine derivative, which is reported to have in-vitro activity similar to that of imatinib and comprises ~15% of the area under the curve (AUC) of the parent compound. Imatinib is extensively distributed into tissues and also shows high plasma protein binding (95%), mostly with albumin and α1-glycoprotein, which can be attributed to a large volume of distribution (435 L) and a long half-life (18 hours) [12]. In addition, imatinib shows a linear absorption profile, in the range 25-1,000 mg of dose after oral administration, and, consequently, AUC also increases proportionally [13] and exhibits body weight dependent clearance ranging from 8-14 L/hr [17].
Imatinib is commercially available in hard gelatin capsules (100 mg) or tablets (100 mg or 400 mg) in United States and European market with 400 mg tablets having advantages over 100 mg formulation in terms of reduced dosing frequency thus better patient compliance. Imatinib is regarded as Gold Standard Pharmacotherapy and the recommended dose for adult patients with Philadelphia chromosome-positive CML (Ph+ CML) in its various phases (chronic, accelerated, and blast crisis) is 400 mg [18,19]. The approved dose for malignant unresectable and metastatic GISTs is 400-600 mg/day [20]. Daily dose ranges from 260-340 mg/m2 in children with CML. The treatment is continued until disease progression or intolerable toxicity.
Orally administered tyrosine kinase inhibitors have significantly transformed the treatment of CML, from a fatal cancer in non-transplanted patients to a long-term condition with a steadily increasing prevalence. However, tyrosine kinase inhibitor is a life-long and expensive therapy. Cost of first-generation imatinib varies from approximately £21,000 per patient per year in the United Kingdom to £57,000 in the United States. The significant cost can have a significant impact on health economies worldwide. The introduction of targeted therapies caused an unprecedented “sticker shock” for providers and payers related to the price of the drug [21]. The financial constraints faced by most health systems today make it necessary for manufacturers of new, expensive drugs to demonstrate value for money [22]. Given the potential for patients with CML to achieve a near normal lifespan, having a drug on the market that is affordable to patients is very important. The current prices of tyrosine kinase inhibitors are high, and generic formulations might reduce healthcare costs [21]. Hence, a new generic imatinib mesylate tablet (400 mg) for once daily administration has been developed by Ranbaxy Laboratories Limited, India. To comply with regulatory requirements for marketing authorization, a study was designed to characterize the pharmacokinetic profile and to assess the bioequivalence of the sponsor’s test formulation (imatinib mesylate 400 mg tablets) with an innovator product (Gleevec 400 mg tablets, Novartis Pharmaceuticals, East Hanover, NJ) under fed conditions, in adult patients of Ph+ CML stabilized on imatinib mesylate 400 mg. In addition, the aim of this study was to monitor the safety profile of Investigational Medicinal Products (IMPs).
Go to : 
Materials and Methods
1. Ethics
Being a multi-center trial, an Investigator from each site submitted the protocol, patient information sheet, informed consent form, patient diary card, and other study documents to the respective Independent Ethics Committee (IEC)/Institutional Review Board (IRB) for review and approval. The protocol was approved by all IEC/IRBs. The study was conducted according to the current version of the Declaration of Helsinki (Seoul 2008 and Brazil 2013) and in compliance with the current ICMR Guidelines for Biomedical Research on Human Patients, Schedule Y (amended version 2013) of Drug and Cosmetics Act, ICH GCP Guidelines and other applicable regulatory guidelines.
2. Patients
A total of 42 adult patients of CML with documented evidence of Ph+ chromosome, meeting the predefined inclusion and exclusion criteria as per the approved protocol were included in the trial/study. Selected patients were already on a stabilized dose of Imatinib 400 mg tablets for at least 30 days prior to administration of test or reference product. Patients included in the study were of either sex (male or female), age ≥ 18 years, female patient with negative pregnancy tests, able to give written informed consent for participation in the trial, having an Eastern Cooperative Oncology Group performance status of 0-2 and an adequate bone marrow, renal and hepatic function. Patients with known hypersensitivity to imatinib mesylate, positive human immunodeficiency virus infection, hepatitis B surface antigen, hepatitis C virus (HCV) and hepatitis A virus (HAV) antibodies, patients in accelerated phase of CML or blast crisis or undergone hematopoietic stem cell transplantation or receiving CYP3A4 family modulators were excluded from the study. Demographic details of patients included in the study are shown in Table 1.
Table 1.
Demographic details of patients
![]()
3. Clinical procedure
This was a multi-center, randomized, open label, two-period, two-treatment, crossover, single dose bioequivalence study, in adult patients of Ph+ CML stabilized with imatinib mesylate 400 mg under fed conditions. Patients were recruited from four sites with site IDs A, B, C, and H. Patients stabilized on imatinib mesylate 400 mg tablets were screened within 10 days prior to period 1 dosing day (day 1). IMP was administered according to the randomization schedule generated using validated SAS software. After overnight fasting for at least 10 hours, patients were served a standardized light (low fat) breakfast/meal (approximately 450 to 550 calories) 30 minutes prior to scheduled dosing time on day 1 (period I) and day 8 (period II). A single oral dose (400 mg) of IMP was administered 30 minutes after serving breakfast/meal, in the sitting position with approximately 240 mL of water at ambient temperature and no food was allowed for at least 4 hours post-dose while water was allowed adlibitum except for 1 hour before and after drug administration. Patients were advised to remain in sitting position for the first 3 hours post-dosing. Two periods were separated by a time period of 7 days during which patients were put on the same imatinib 400 mg tablets on which they were stabilized prior to period 1. A total of 42 venous blood samples were collected from each patient (except for withdrawn patients) during the study course. The venous blood samples were withdrawn at pre-dose (duplicate pre-dose samples) and at 0.500, 1.000, 1.500, 2.000, 2.500, 3.000, 3.500, 4.000, 4.500, 5.000, 6.000, 7.000, 8.000, 9.000, 10.000, 12.000, 16.000, 20.000, and 24.000 hours post-dose on day 01 and day 08. Collected blood samples were centrifuged at 3,000±100 rcf for 5 minutes at room temperature for separation of plasma. Blood samples were kept in an ice cold water bath before centrifugation and during separation of plasma. The separated plasma was transferred to pre-labeled polypropylene tubes in two aliquots and stored in upright position in a box containing dry ice or in a freezer at a temperature of –65±10°C for interim storage until shipment to the analytical site. During shipment the samples were packed in boxes containing asn adequate amount of dry ice, no interruption of the freeze cycle was allowed. Shipment was done separately for each aliquot and stored at –65±10°C at a bio-analytical facility. Analysis was initiated once samples were available from any of the multi-center sites.
4. Bio-analytical procedure (liquid chromatography–tandem mass spectrometry)
In house liquid chromatography-tandem mass spectrometry method was developed and validated for quantitative analysis of Imatinib in plasma samples. The developed method was validated for various validation parameters (Table 2) according to the guidelines [23]. Analytical procedure was performed at Lambda Therapeutics Research Limited (Gujrat, India) using an instrument equipped with a MS/MS component (Quattro Premier XE, Waters, Manchester, UK), sample manager with column oven (Aquity SM, Waters), and binary solvent manager (Acquity BSM, Waters). The mobile phase consisted of methanol and ammonium formate buffer (5 mM, pH 7.0) at a ratio of 75:25 v/v. Chromatographic separation was achieved using a C18 column (ACE, 5μ, 150x4.6 mm) at the flow rate of 1 mL/min and injection volume (10 μL) in full loop mode. Mass-spectrometric detection was performed using an electrospray-ionization (ESI) technique and the ESI source was kept in positive ionization mode. MRM transitions for imatinib (analyte) and Imatinib-d8 (internal standard) were m/z 494.3 > 394.16 and 502.44 > 394.21, respect-ively. Data acquisition and evaluation was performed using Mass Lynx software ver. 4.1 (Waters).
Table 2.
Summary of method validation parameters
![]()
5. Pharmacokinetic analysis
The pharmacokinetic parameters evaluated for imatinib were maximum plasma concentration (Cmax), area under the plasma concentration versus time curve from time zero to truncated time at 24 hours (AUC0-24), time to reach maximum plasma concentration (Tmax). Cmax and Tmax were calculated directly from the plasma concentration time profiles of the patients. AUC0-24 was calculated using the linear trapezoidal method. Pharmacokinetic parameters were calculated for imatinib using a non-compartmental model using WinNonlin Professional Software ver. 5.3 (Pharsight Corp., Mountain View, CA).
6. Statistical analysis
Statistical comparison of pharmacokinetic parameters for both formulations (test and reference) was performed using PROC GLM and PROC MIXED of SAS ver. 9.3 (SAS Institute Inc., Cary, NC) for assessment of the bioequivalence of the formulations.
Data of patients from sites with less than five evaluable patients were pooled in the previous site. Considering that site ‘B’ and site ‘C’ had less than five evaluable patients, the data from both sites were pooled in site ‘A’ data.
The center effect was carried out using the PROC GLM of SAS ver. 9.3 (SAS Institute Inc.). Comparison of the pharmacokinetic parameters was performed using PROC MIXED of SAS ver. 9.3 (SAS Institute Inc.).
Analysis of variance (ANOVA) using type III sum of squares for imatinib was performed for the ln-transformed pharmacokinetic parameters Cmax and AUC0-24. ANOVA model included the terms for center, sequence, center by sequence, period (within center), formulation, formulation by center and patient (within center by sequence). The sequence effect was tested at the 10% level of significance using the patient nested within center sequence effect as the error term. Other remaining effects were tested at the 5% level of significance against the residual error (mean square error) from the ANOVA model as the error term. Each ANOVA included the calculation of least-squares means, the difference between the adjusted formulation means and the standard error associated with the difference. The above analyses were performed using an appropriate SAS procedure.
Consistent with two one-sided tests for bioequivalence, 90% confidence intervals for the ratio of geometric least squares means (LSM) between test and reference formulations were calculated for ln-transformed pharmacokinetic parameters Cmax and AUC0-24 of imatinib. In addition, the power of the study was computed and reported for ln-transformed pharmacokinetic parameters Cmax and AUC0-24 for detection of a 20% difference between test and reference formulations. In addition, ratio of geometric least squares means of test and reference formulations was reported for ln-transformed pharmacokinetic parameters Cmax and AUC0-24 of imatinib. Intra-patient and inter-patient variability was also computed for ln-transformed pharmacokinetic parameters Cmax and AUC0-24.
Test and reference product were considered bioequivalent if the 90% confidence interval for the ratio of test product and reference product for the ln-transformed pharmacokinetic parameters Cmax and AUC0-24 of imatinib were within the acceptance range of 80.00%-125.00%.
7. Safety assessment
Medical history, physical examination and vital signs, body measurements, immunological Tests (hepatitis B surface antigen, HAV [IgM], and HCV antibody), pregnancy test for female patients, electrocardiography and echocardiography evaluation, chest X-ray, standard clinical laboratory evaluations (hematology, blood chemistry, urinalysis), and adverse event (AE) monitoring were performed as a part of safety evaluations. In addition, participants were encouraged to report any undesirable side effects such as nausea, vomiting, or headache. Any spontaneously reported AEs were also recorded and managed. In addition, pre- and post-study laboratory parameters were evaluated to establish the safety of formulation.
Go to :
Results
1. Bio-analytical procedure
A summary of the validation parameters along with observed range and acceptable deviation as per the guidelines is shown in Table 2 [23]. An in-house developed and validated bioanalytical procedure was found to comply with all acceptability limits and therefore suitably applied for the quantitative estimations of plasma samples.
2. Patients and drug accountability record
The bioequivalence study was conducted in 42 adult patients of Ph+ CML stabilized with Imatinib mesylate 400 mg tablets, after administration of a single dose under fed conditions. A summary of the demographic data of the participants is shown in Table 1.
3. Pharmacokinetics and statistics
The mean plasma drug concentration versus time profiles achieved after a single oral dose of imatinib (400 mg of test and reference formulations) are shown in Fig. 1. A non-compartment model was used to assess the pharmacokinetic parameters. The mean pharmacokinetic parameters, Cmax, Tmax, and AUC0-24 for test and reference product are shown in Table 3. The statistics of pharmacokinetic parameters, i.e., mean co-efficient of variation, ratio of LSM of the test and the reference product, 90% confidence intervals, power for ln-transformed pharmacokinetic parameters are shown in Table 3. Two patients (A-14 and A-04) did not complete the study and were withdrawn from the trial due to AEs in period I and period II, respectively. As per the protocol requirement, plasma samples of withdrawn patients were analyzed but these patients were not included in pharmacokinetic and statistical analysis.
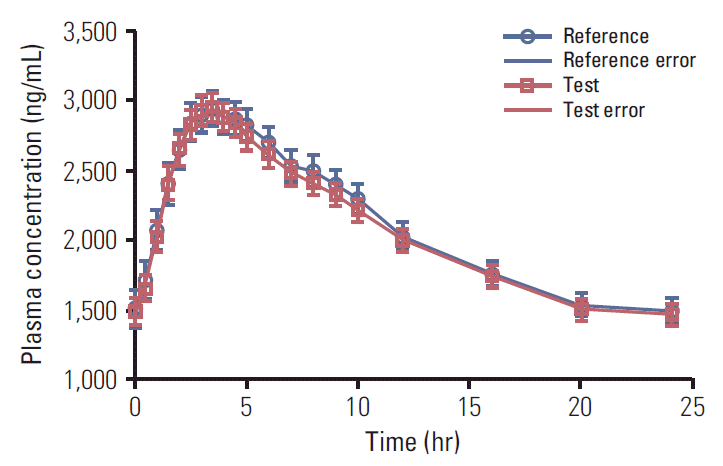 | Fig. 1.Plasma drug concentration verses time profiles of test and reference product of imatinib mesylate 400 mg tablets in Philadelphia chromosome positive chronic myeloid leukemia patients (n=40).
|
Table 3.
Pharmacokinetic parameters and relative bioavailability of test and reference formulation of imatinib under fed condition in Ph+ CML patients (n=40)
| Parameter |
Untransformed data |
Geometric least squares means |
90% Confidence interval (test vs. reference) | Power (%) | |||
|---|---|---|---|---|---|---|---|
| Test | Reference | Test | Reference | Ratio (test/reference, %) | |||
| Tmax (hr)a) | 3.250 (1.500-6.000) | 3.500 (1.500-8.000) | - | - | - | - | - |
| Cmax (ng/mL) | 3,101.911±658.9118 | 3,155.945±790.9928 | - | - | - | - | - |
| AUC0-24 (ng·hr/mL) | 48,977.355±12,453.3736 | 49,870.552±15,003.0949 | - | - | - | - | - |
| lnCmax | - | - | 3,026.238 | 3,057.880 | 99.0 | 94.03-104.16 | 100 |
| lnAUC0-24 | - | - | 47,381.318 | 47,761.108 | 99.2 | 94.72-103.90 | 100 |
![]()
Descriptive statistics were calculated and reported for primary and secondary pharmacokinetic parameters for imatinib. The mean Cmax, Tmax, and AUC0-24 for the test product of imatinib were 3,101.911±658.9118 ng/mL, 3.425±1.0893 hours and 48,977.355±12,453.3736 ng·hr/mL, respectively, and 3,155.945±790.9928 ng/mL, 3.838±1.4736 hours, 49,870.552±15,003.0949 ng·hr/mL, respectively, for the reference product. The test/reference ratios for ln-transformed data for the pharmacokinetic parameters Cmax and AUC0-24 were 99.0 and 99.2, respectively, while 90% confidence intervals for Cmax and AUC0-24 were in the range of 94.03%-104.16% and 94.72%-103.90%, respectively (Table 3). Intrasubject variability was expressed in terms of percentage coefficient of variance and reported as 13.6% and 12.3%, respectively, for Cmax and AUC0-24.
Probability (p-value) for lnCmax and lnAUC0-24 was estimated by ANOVA for various effects. In statistical evaluation, center, center*sequence, formulation and period (center) effects was considered statistically significant if p-value < 0.05 while sequence effect was considered statistically significant if p < 0.01. Based on the results (Table 4), it was concluded that center, sequence, and center*sequence, formulation and period (center) effects were statistically non-significant for ln-transformed pharmacokinetic parameters Cmax and AUC0-24 for imatinib.
Table 4.
ANOVA p-values, intra- and inter-patient CV for imatinib
![]()
4. Safety evaluation
All patients (except for withdrawn patients, A-04 and A-14) were subjected to a single oral dose according to the randomization schedule and received both IMPs.
The AEs observed during investigation periods are shown in Tables 5 and 6. Eleven AEs were reported by nine patients. Two AEs were reported in period I, one AE was reported on the day of check-in for period II, three AEs were reported in period II, and five AEs were reported during post-study safety assessment. The analysis of AEs was performed treatment-wise. Four patients out of 41 patients (9.76%) in the reference group reported four AEs. The causality assessment of three AEs was possible while one AE was unlikely. Five patients out of 41 patients (12.20%) in the test group reported seven AEs. The causality assessment of all seven AEs was unlikely.
Table 5.
Day-wise bifurcation based on the onset of adverse events
| Visit/Day | No. of adverse events |
|---|---|
| Day 1 (period 1) | 2 |
| Day 7 (check-in period 2) | 1 |
| Day 8 (period 2) | 3 |
| End of study assessment | 5 |
| Total | 11 |
![]()
Table 6.
Summary of AEs
![]()
Go to :
Discussion
Cancer is not only regarded as a disease, as it instills terror, anxiety, and is considered an evil and unbeatable killer. Anticancer drugs are usually of high cost and even when most approved cancer drugs provide little gain for patients, drives much of the research as patients are anxious and willing to pay whatever companies charge. Regulatory agencies are keen to introduce new anticancer drugs on the market. A review of drugs for solid cancers approved by the European Medicines Agency in its first 10 years found that, overall, new oncology drugs improved survival by a mean and median of 1.5 and 1.2 months, respectively. The 71 drugs approved by the FDA from 2002 to 2014 for solid tumors have resulted in median gains in progression-free and overall survival of only 2.5 and 2.1 months, respectively [24].
Improving economy and health care facilities in developing countries like India has led to increased average life span of an individual and the disease scenario has switched from communicable to non-communicable ailments. Cancer is one of the deadliest diseases seen in the modern world. World Health Organization (WHO) estimates that if the current rate of cancer remains unchanged, new cases of cancer will increase from 14 million cases (2012) to 22 million cases within two decades (WHO report 2014). Unfortunately, India has one of the largest out-of-pocket health care expenditures worldwide; despite offering economical services in comparison to United Kingdom and United States, it is unaffordable to a large group of the population due to poor income. Over the years, increasing healthcare costs (particularly in cancer chemotherapy) has led to development of the concept of control over drug pricing. Thus, providing a low cost commercially available formulation is the most promising approach, which can be achieved by launching generic medicine on the market. A generic medicine is an alternative to a "reference medicine" that has already been approved and authorized; it contains the same active substance(s), used in equal dose(s) for treatment of the same disease(s) as the reference medicine. Generic medicines are frequently being used in several countries, considering pharmacoeconomics, and physicians are showing interest in effective alternatives available at a low price over high priced innovator products.
A similar effect can be achieved by launching generic drugs at a low cost; however, they should demonstrate bioequivalence with innovator products. US FDA defined it as “the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study.” Bioequivalence is a pharmacokinetics term used for evaluation of the anticipated in-vivo biological equivalence of two proprietary preparations of a drug.
Imatinib is a cytotoxic drug; hence, cancer patients were considered as an appropriate population for conduct of study to avoid unnecessary exposure to healthy volunteers. Imatinib is indicated for treatment of patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase as well for treatment of patients with Kit (CD117) positive unresectable and/or metastatic malignant GISTs. The study was conducted with a single dose of the highest recommended dosage strength and plasma samples were efficiently quantified for Imatinib using a developed and validated bio-analytical method which was sensitive enough (LLOQ, 5.021 ng/mL) for quantification of the samples.
As shown in Fig. 1, the mean concentration vs. time profile curve was comparable between test and reference formulations. Test/reference ratios for ln-transformed data for the pharmacokinetic parameters Cmax and AUC0-24 were close to unity. The ratio and 90% confidence interval of geometric least squares means of test product and reference product for ln-transformed Cmax and AUC0-24 were 99.0% (94.03%-104.16%) and 99.2% (94.72%-103.90%), respectively, which are within the acceptance limit of 80.00%-125.00%, set for bioequivalence. Hence, the test product met the bioequivalence criteria with respect to the rate and extent of absorption of Imatinib under fed conditions as per US FDA criteria.
ANOVA p-values for log transformed Cmax and AUC0-24 were determined in two ways, first, formulation*center effect was evaluated to check the pool-ability of the data. Formulation*center effect was found to be statistically non-significant at a 5% level, hence this interaction term (formulation*center) was dropped from the ANOVA model. A statistical analysis was re-performed by excluding this interaction term and ANOVA p-values were determined. The sequence effect was tested at the 10% level of significance using the subjects nested within sequence mean square as the error term. All other main effects were tested at the 5% level of significance against the residual error (mean square error) from the ANOVA model as the error term. Center, sequence, center*sequence, formulation, period (center) effect for ln-transformed Cmax and AUC0-24 were insignificant. The power of the test for both lnCmax and lnAUC0-24 was 100%, within the required limit of ≥ 80%. The overall intra subject variability for lnCmax and lnAUC0-24 were 13.2% and 12.6%, respectively. Intra-patient variability was low (< 30%) indicating that sample size and study design for the current bioequivalence study is reasonable enough to provide adequate statistical power to comply with FDA bioequivalence requirement.
Pre-enrollment laboratory and vital parameters for all patients were within clinically acceptable ranges for enrollment in the study. Blood cell count, hemoglobin, serum bilirubin, alanine transaminase, aspartate aminotransferase, alkaline phosphatase, and serum creatinine were performed before dosing on day 1 (or one day prior to period 1 dosing) and before dosing on day 8 (or one day prior to period 2 dosing). Laboratory assessments for all patients were within clinically acceptable ranges, except for patient A-04. The laboratory abnormality was recorded as an AE. Similarly, laboratory parameter evaluation performed after period 2 (as safety follow up) showed that all patients were within clinically acceptable ranges, except for patient A-07, A-11, and C-01.
Out of the eleven AEs reported, nine AEs were mild in nature and two AEs were moderate. The causality assessment was judged as possible for three and as unlikely for eight AEs. The most frequently reported AE was fever (four cases reported). There were no deaths during the conduct of study; however, one serious adverse event (SAE) (viral fever in patient A-14) was reported. Ten of the eleven AEs recovered without sequelae and outcome of one AE was unknown. Patient A-14 was withdrawn from the study due to SAE and was followed up until resolution of SAE. The causality assessment of the SAE to the study drug was judged to be unlikely. Both IMPs were well tolerated by Ph+ CML patients. Thus, it can be concluded that both the test and reference products can be safely administered.
Go to :
Conclusion
In conclusion, in a bioequivalence study for imatinib (Gleevec vs. imatinib mesylate 400 mg tablets) conducted in Ph+ CML patients, stabilized on imatinib mesylate tablets 400 mg, test and reference formulations of imatinib met the regulatory criteria for bioequivalence based on the rate and extent of absorption. Based on the finding of this study, it can be concluded that the product is bioequivalent and safe, thus suggesting the clinical application of the test product as an alternative to Gleevec.
Go to :
 XML Download
XML Download