

 PDF
PDF Citation
Citation Print
Print


Introduction
Bladder cancer is a common human malignancy [1]. Despite development of conventional therapies in recent decades, finding new molecular targets and development of new targeted therapy for achievement of long-term patient survival is still important.
TRIM29 (ATDC), a member of TRIM family proteins, has been shown to regulate cell growth and play important roles in human diseases [2]. Several TRIM family proteins, including TRIM8 and TRIM40, regulated nuclear factor κB (NF-κB) signaling activation [3,4]. TRIM29 was first found to be responsible for ataxia-telangiectasia, a genetic disorder, and recent studies reported involvement of TRIM29 in human cancers including gastric cancer, pancreatic cancer, and non-small cell lung cancer [2,5]. In non-small cell lung cancer, overexpression of TRIM29 promoted cancer cell proliferation and invasion through NF-κB and ERK signaling [5]. TRIM29 promotes pancreatic cancer cell proliferation in vitro and tumor growth and metastasis in vivo by upregulation of β-catenin signaling. Downregulation of TRIM29 was also reported in breast cancer and prostate cancer [6,7]. However, the clinical significance and biological functions of TRIM29 in bladder cancers remain unexplored.
In the current study, we examined clinical significance of TRIM29 in bladder cancer using immunohistochemistry. We also examined the effect of overexpression and downregulation of TRIM29 expression in bladder cancer cell lines on cell proliferation, invasion, cell cycle progression, and apoptosis, as well as the molecular signaling pathways underlying the biological effect of TRIM29.
Materials and Methods
1. Clinical specimens
The protocol of the current study was approved by the Review Board of Shengjing Hospital. Clinical tissues were collected from 102 patients diagnosed with bladder cancer in the Affiliated First Hospital and Shengjing Hospital of China Medical University since 2010 to 2013. The histological diagnosis and tumor grade were evaluated by a pathologist according to the World Health Organization (WHO) classification guidelines. Tumors were classified according to Ta, T1, T2, T3, and T4 based on WHO guidelines.
2. Immunohistochemistry
Tumor specimens were fixed with 10% neutral formalin, and 4-μm-thick paraffin sections were made. Immunostaining was performed using the S-P staining kit from MaiXin (Ultrasensitive, MaiXin, Fuzhou, China). After antigen retrieval in citrate buffer (pH 6.0) for 2 minutes in an autoclave, 0.3% hydrogen peroxide was applied for 15 minutes, followed by incubation of the sections with goat serum. The sections were then incubated with TRIM29 rabbit polyclonal antibody at 4°C overnight (1:300, Proteintech, Rosemond, IL). Incubation with primary antibody was performed at room temperature for 2 hours, followed by incubation with biotinylated goat anti-rabbit serum IgG after washing in phosphate buffered saline (PBS), and then incubation with horseradish peroxidase (HRP) conjugated streptavidinbiotin. A DAB kit (MaiXin) was used for staining. Counterstaining with hematoxylin was performed and the sections were dehydrated in ethanol before mounting.
All slides were examined randomly by two independent pathologists. Five views were examined per slide. Immunostaining of TRIM29 was scored on a semiquantitative scale by evaluating the intensity and percentage of cells showing positive staining. Cytoplasmic and membrane staining was considered as positive immunostaining. The intensity of TRIM29 cytoplasmic staining was also scored as 0 (none), 1 (weak), and 2 (marked). Percentage scores were assigned as follows: 1, 1%-25%; 2, 26%-50%; 3, 51%-75%; and 4, 76%-100%. The scores were multiplied to give a final score of 0 to 8 and the total expression of TRIM29 was determined as either negative/weak expression (score < 4) or overexpression (score ≥ 4).
3. Cell culture and transfection
SV-HUC-1, BIU-87, 5637, and T24 cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA), which were cultured in 1640 medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Invitrogen). Cells were passaged every 2 days with trypsin.
DharmaFECT1 (Dharmacon, Lafayette, CO) was used for siRNA knockdown. ONTARGETplus siRNA for TRIM29 was also obtained from Dharmacon. Non-targeting siRNAa were used as negative control.
pCMV6-TRIM29 plasmid was obtained from Origene (Rockville, MD). Attractene Transfection was used for transfection of plasmid (Qiagen, Hilden, Germany). pCMV6 was used as control.
BAY 11-7082 (Sigma, St. Louis, MO) was used as NF-κB inhibitor. Protein kinase C (PKC) inhibitor staurosporine was purchased from Beyotime Biotechnology (Shanghai, China).
4. Western blot analysis
Total protein was extracted using Pierce lysis buffer (Pierce, Rockford, IL). Protein quantification was performed using the Bradford method. A 50 μg sample of protein was added to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Bradford, MA). Incubation of primary antibodies was performed overnight at 4°C. Antibody dilution was as follows: TRIM29 (1:800, Proteintech), p-PKC, p-IκB, cyclin D1, cyclin E, Bcl-2 (1:1,000, Cell Signaling, Boston, MA), and mouse monoclonal antibody against β-actin (1:2,000, Santa Cruz Biotechnology, Santa Cruz, CA). This was followed by incubation with HRP-coupled anti-mouse or rabbit IgG antibody (1:1,000, Cell Signaling Technology, Danvers, MA) at 37°C for 2 hours. Target proteins on polyvinylidene difluoride membrane were visualized using a Pierce ECL kit and captured using a DNR BioImaging System (DNR, Jerusalem, Israel).
5. Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (PCR) was performed using a SYBR Green master mix kit (Applied Biosystems, Foster City, CA). PCR was performed using a 7500 Real-Time PCR System (Applied Biosystems). β-Actin was used as the endogenous control. The relative levels of gene expression were represented as ΔCt=Ct gene–Ct reference, and the fold change of gene expression was calculated using the 2–ΔΔCt method. Experiments were performed in triplicate. The primer sequences are as follows: TRIM29 forward, 5'-GCACCGGACACCATGAAGA-3'; TRIM29 reverse, 5'-GGAGACGAGGGCTGGTATGA-3'; cyclin D forward, 5'-TGGAGGTCTGCGAGGAACA-3'; cyclin D reverse, 5'-TTCATCTTAGAGGCCACGAACAT-3'; cyclin E forward, 5'-AGC-CAGCCTTGGGACAATAAT-3'; cyclin E reverse, 5'-GAGC-CTCTGGATGGTGCAAT-3'; Bcl-2 forward, 5'-ACGGTGGTGGAGGAGCTCTT-3'; Bcl-2 reverse, 5'-CGGTTGACGCT-CTCCACAC-3'; β-actin forward, 5'-ATAGCACAGCCTGGATAGCAACGTAC-3'; β-actin reverse, 5'-CACCTTCTACAATGAGCTGCGTGTG-3'.
6. Cell Counting Kit-8 cell proliferation test
Cell proliferation speed was analyzed using a Cell Counting Kit-8 (CCK-8) kit (Dojindo, Gaithersburg, MD) according to the manufacturer’s protocol. Briefly, 48 hours after transient plasmid or siRNA transfection, cells were seeded at approximately 6×103 in 96-well plates. Each day the cells were treated with 10 μL CCK-8 solution for 4 hours, followed by measurement at 450 nm using a microplate reader.
7. Reporter assay
NF-κB reporter plasmid was purchased form Beyotime (0.2 μg, Beyotime Biotechnology) and Renilla luciferase reporter plasmid was obtained from Promega (Madison, WI). Both plasmids were transfected into bladder cancer cells using Attractene reagent (Qiagen). Approximately 30 hours after transfection, a dual luciferase reporter kit (Promega) was added to cells and luciferase activity was examined.
8. Cell cycle analysis
Forty-eight hours after transfection, cells were harvested and fixed using 1% paraformaldehyde. The cells were then washed with PBS and stained in 5 mg/mL propidium iodide (PI) for 30 minutes at room temperature. Flow cytometry was performed using a BD FACS Calibur flow cytometer system (Becton Dickinson, Franklin Lakes, NJ).
9. Cell apoptosis detection
Cell apoptosis detection was performed with Annexin V/PI double staining. Forty-eight hours after transfection, cells were harvested, washed in chilled PBS. The cells were then re-suspended in 250 μL of binding buffer. Annexin V/fluorescein isothiocyanate solution and PI solution were added to the cell suspension. Following incubation for 30 minutes, the cells were analyzed using a BD FACS Calibur flow cytometer (Becton Dickinson).
Results
1. Expression of TRIM29 in normal and bladder cancer tissues
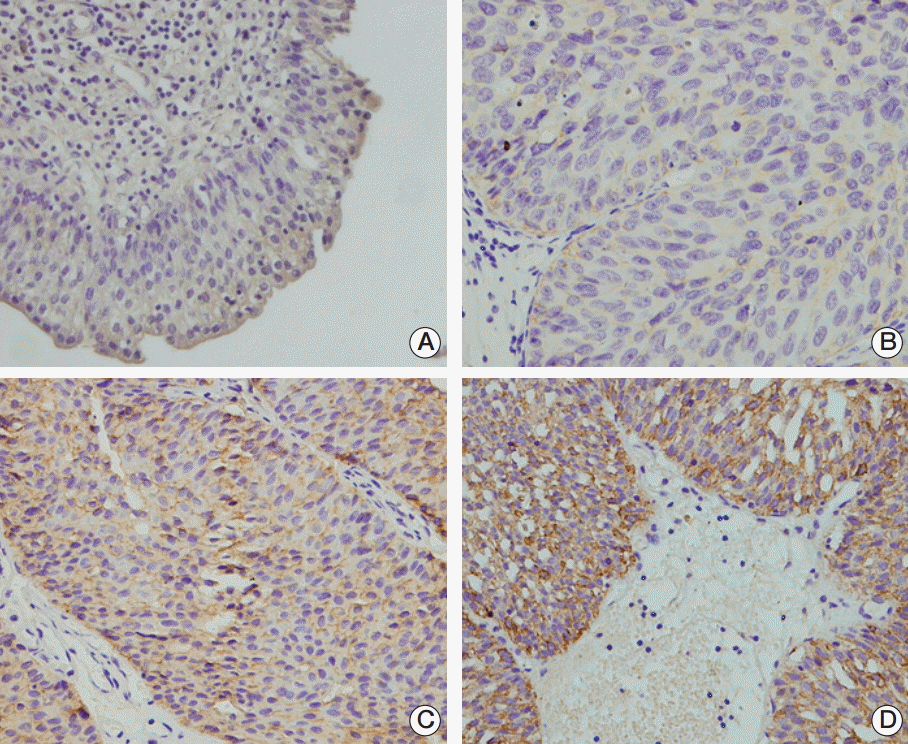
To examine the TRIM29 protein expression pattern in bladder cancer, a panel of 102 primary bladder cancer samples was analyzed by immunohistochemistry. TRIM29 staining was observed in the cytoplasmic and membrane compartment of bladder cancer cells and cytoplasmic TRIM29 staining was considered positive. Weak/negative TRIM29 staining was observed in normal bladder tissue (Fig. 1A). Overexpression of TRIM29 was observed in 45 out of 102 bladder cancer tissues (44.1%) (Fig. 1B-D). We also examined the relationship between TRIM29 expression and clinical factors. No statistical differences were observed between TRIM29 overexpression and the characteristics of age, sex, and tumor grade (Table 1). However, TRIM29 overexpression showed correlation with local invasion status (Ta-T1 vs. T2-T4, p=0.009).
2. Expression of TRIM29 in human bladder cancer cell lines and its role in cell proliferation
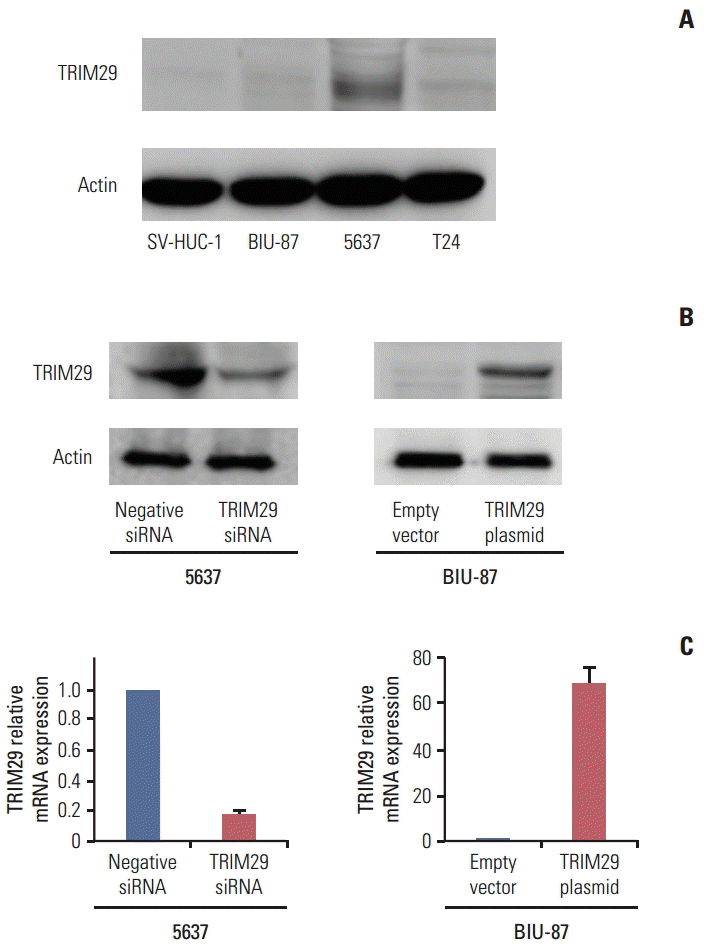
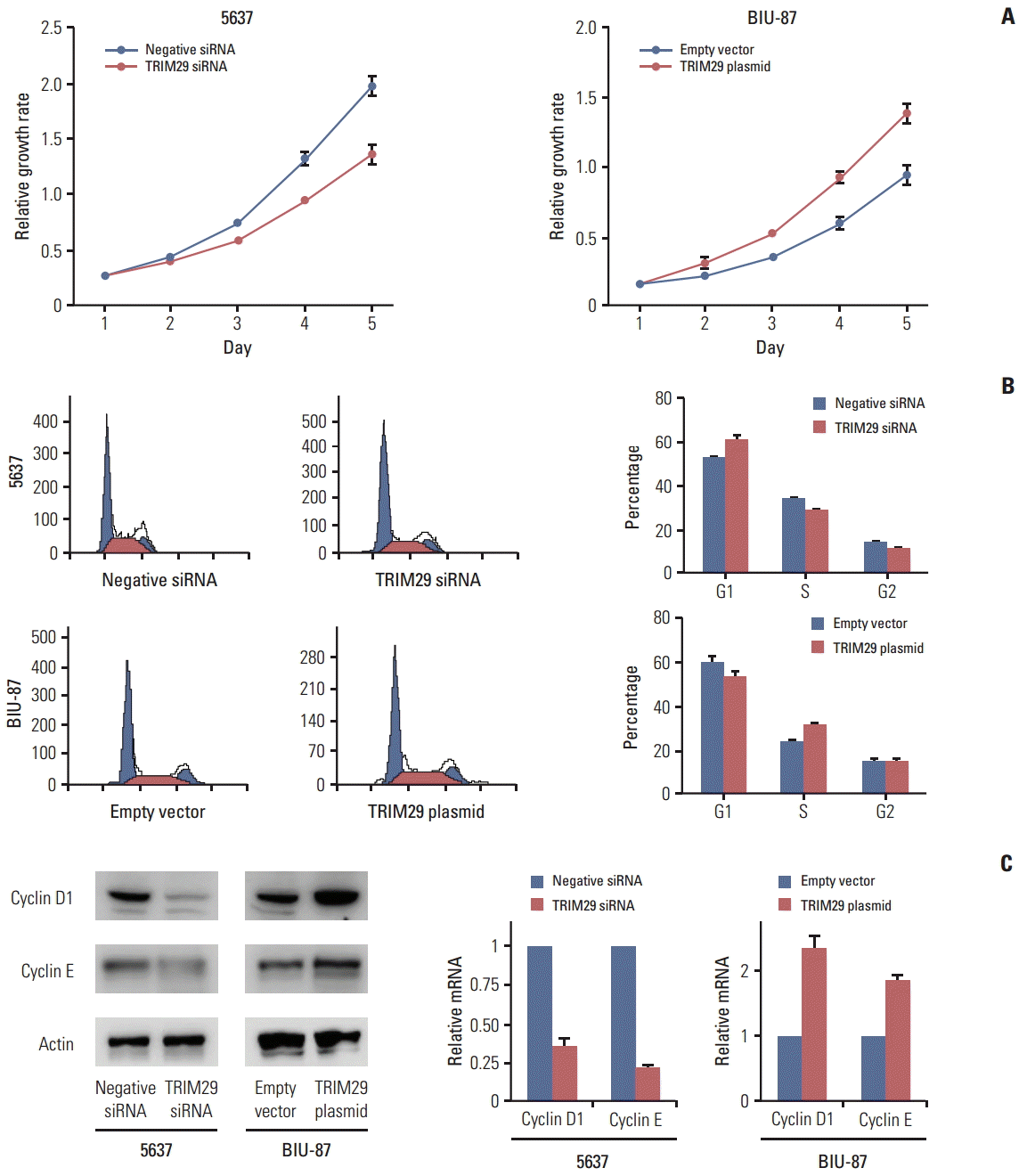
TRIM29 expression level was detected in three bladder cancer cell lines and normal cell line SV-HUC-1 by western blot. Relatively high TRIM29 protein expression was found in the 5637 cell line and relatively low TRIM29 level was observed in BIU-87 and SV-HUC-1 cell lines (Fig. 2A). To examine its biological function, we employed TRIM29 specific siRNAs in the 5637 cell line. TRIM29 plasmid transfection was performed in the BIU-87 cell line. We then confirmed the TRIM29 siRNA knockdown and plasmid transfection efficiency (Fig. 2B and C). The effect of TRIM29 on bladder cancer proliferation was examined using a CCK-8 kit. The results showed that TRIM29 transfection promoted cell growth (day 5; empty vector vs. TRIM29 plasmid, 0.931±0.037 vs. 1.381±0.057; p < 0.05) and its depletion inhibited cell proliferation (day 5; negative siRNA vs. TRIM29 siRNA, 1.928±0.085 vs. 1.301±0.081; p < 0.05) (Fig. 3A).
3. TRIM29 facilitates cell cycle transition in bladder cancer cells
In cell cycle analysis, we found that G1 phase percentage was downregulated after TRIM29 transfection in BIU-87 cells (EV vs. TRIM29 plasmid, 60.6±0.8 vs. 54.3±1.2) and increased in 5637 cells treated with siRNA (control vs. TRIM29 siRNA, 52.7±1.3 vs. 61.5±2.1) (Fig. 3B). The S phase percentage was upregulated in BIU-87 cells with TRIM29 overexpression (EV vs. TRIM29, 24.2±0.5 vs. 30.6±0.7) and decreased in TRIM29 depleted cells (control vs. TRIM29 siRNA, 33.6±1.2 vs. 28.1±0.9).
We also found that TRIM29 depletion led to downregulated cyclin D1 and cyclin E in the 5637 cell line at both mRNA and protein levels. TRIM29 overexpression increased cyclin D1 and cyclin E in the BIU-87 cell line (Fig. 3C). The above results indicated that TRIM29 regulates cell cycle related proteins and facilitates cell cycle progression.
4. TRIM29 inhibits apoptosis and upregulates Bcl-2 expression
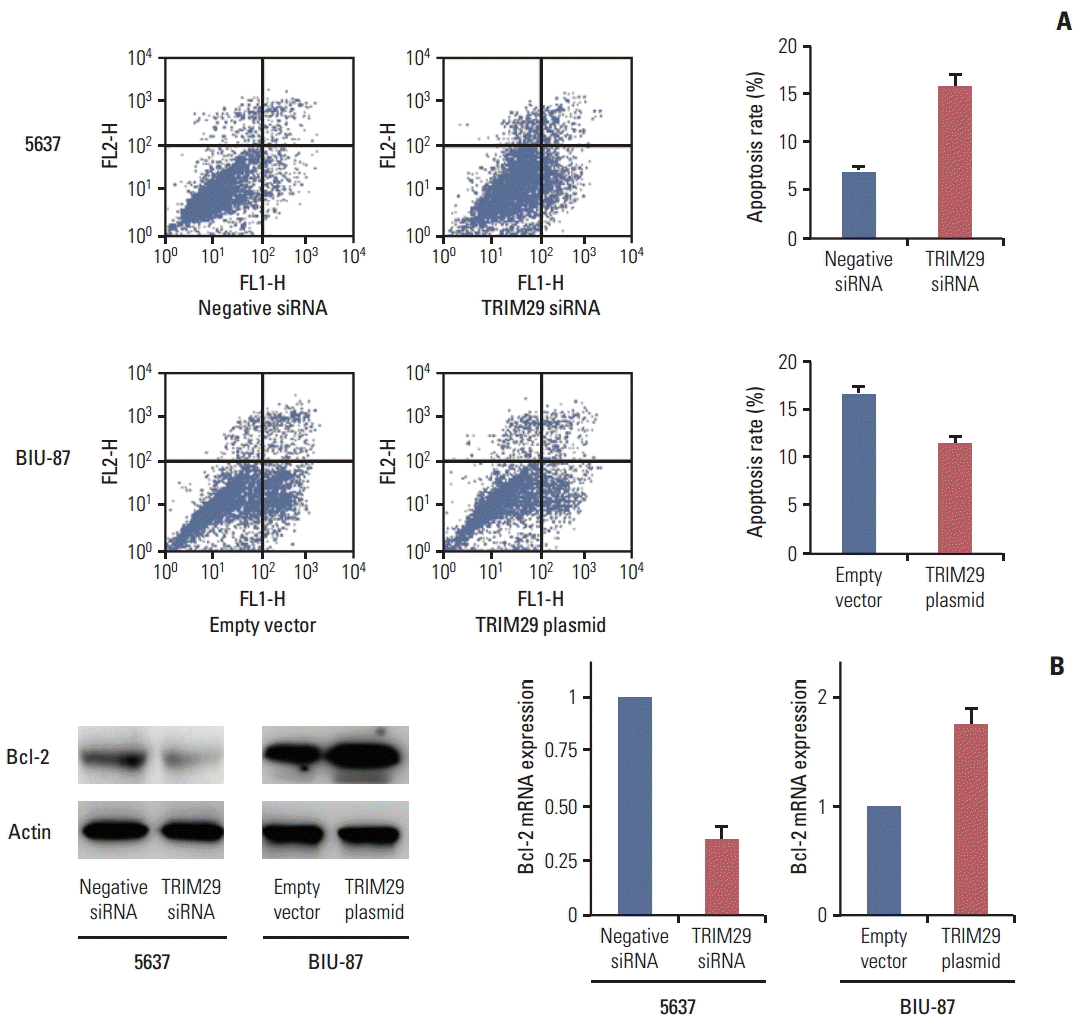
To characterize the effect of TRIM29 on cisplatin-mediated apoptosis, an Annexin V kit was used in 5637 and BIU-87 cells with cisplatin treatment (10 μg/mL, 24 hours). As shown in Fig. 4A, a significantly higher percentage of early and late apoptosis was observed in 5637 cells with TRIM29 depletion compared with negative controls. TRIM29 overexpression in BIU-87 cells inhibited apoptotic percentage, demonstrating that TRIM29 overexpression confers cisplatin resistance of bladder cancer cells. We then checked expression of Bcl-2 protein, which was closely related to apoptosis regulation. As shown in Fig. 4B, TRIM29 siRNA decreased Bcl-2 expression while TRIM29 overexpression upregulated Bcl-2 level in the BIU-87 cell line.
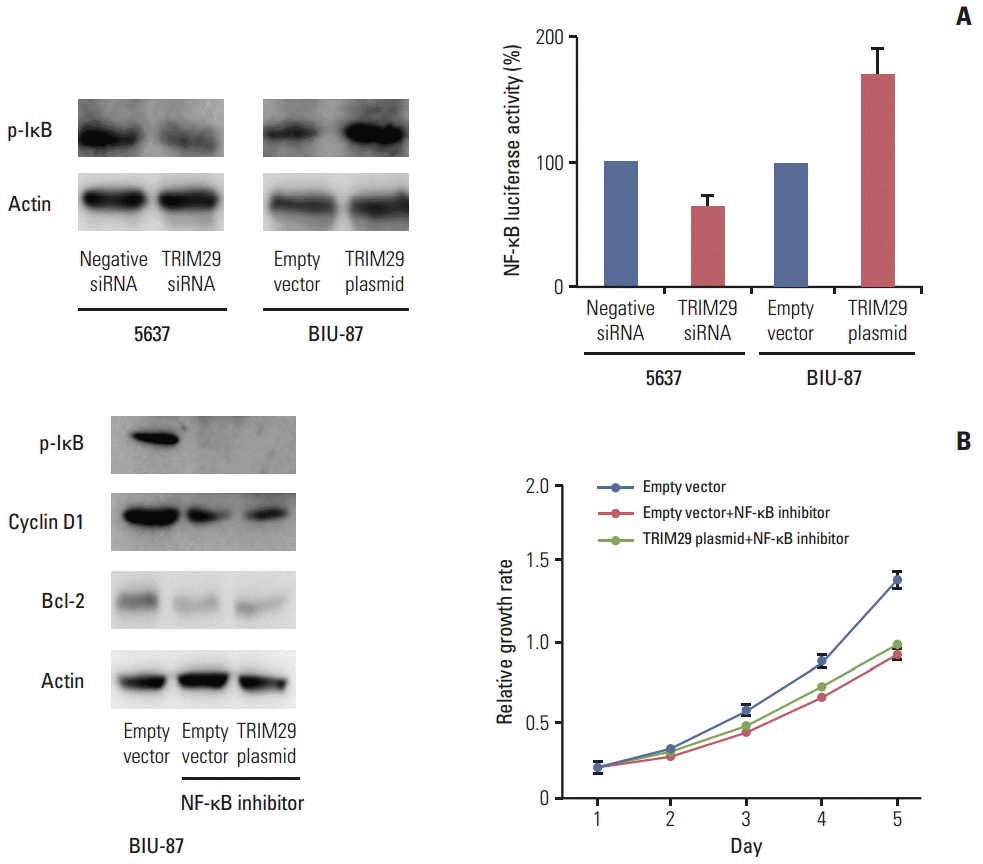
5. TRIM29 regulates cell cycle and apoptosis through NF-κB signaling
Involvement of TRIM29 in NF-κB signaling has been reported. We wondered whether this is also the case in bladder cancer cells. Using a luciferase reporter assay, we showed that TRIM29 transfection increased the level of NF-κB reporter activity and its depletion decreased NF-κB reporter activity. In addition, western blot analysis showed that p-IκB expression was upregulated after TRIM29 overexpression and downregulated after TRIM29 depletion (Fig. 5A). The above results suggested that the effect of TRIM29 on NF-κB may induce cell cycle progression and apoptosis inhibition. To validate the involvement of NF-κB in TRIM29-regulated cell proliferation and apoptosis, we employed Bay 11-7082, which inhibited IkB phosphorylation and NF-κB activation in TRIM29-overexpressed BIU-87 cells. Cells were treated at 5 μM concentration for 10 hours. As shown in Fig. 5B, NF-κB inhibitor blocked the expression of cyclin D1 and Bcl-2 induced by TRIM29. The inhibitor also blocked the effect of TRIM29 on cell proliferation.
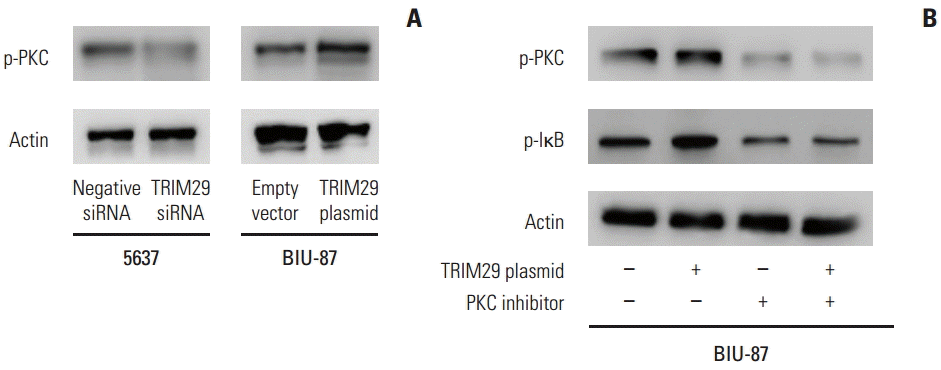
6. TRIM29 regulates the NF-κB pathway through PKC activation
Further analysis was performed to examine the mechanism of TRIM29 on NF-κB signaling. PKC was reported to activate NF-κB signaling in various cancers. Western blot analysis showed that PKC phosphorylation was upregulated after TRIM29 overexpression and downregulated after TRIM29 depletion, suggesting involvement of PKC in NF-κB activation induced by TRIM29 (Fig. 6). We also employed PKC inhibitor staurosporine (10 nM, 12 hours) in TRIM29 transfected BIU-87 cells. The results showed that staurosporine strongly blocked the role of TRIM29 on p-IκB in BIU-87 cells.
Discussion
As one member of the TRIM protein family, overexpression of TRIM29 has been reported in various human cancers [2,5,8]. A previous study identified TRIM29 as a potential oncoprotein in non-small cell lung cancer which regulates cancer cell proliferation and invasion [5]. A recent study reported that TRIM29 was upregulated in colorectal cancer tissues and showed correlation with poor patient prognosis [8]. These studies indicate TRIM29 as a potential oncoprotein in human cancers. However, its expression pattern, biological function, and molecular mechanism in bladder cancer were not clear. In this study, we found elevated TRIM29 expression in bladder cancer tissues and bladder cancer cell lines. A correlation was observed between upregulation of TRIM29 and primary tumor status (invading depth). Expression rate of TRIM29 was higher in T2-T4 tumors than in T1 tumors, suggesting that TRIM29 plays a role in bladder cancer progression.
In this study, we employed siRNAs to downregulate endogenous TRIM29 in 5637 cells which show high endogenous TRIM29 expression and overexpressed TRIM29 in BIU-87 cells. We found that depletion of TRIM29 led to significantly suppressed proliferation while overexpression of TRIM29 promoted proliferation, which was in accordance with previous studies. Cell cycle analysis showed that in the 5637 cell line, depletion of TRIM29 led to inhibited cell cycle transition at G1/S point and transfection of TRIM29 in BIU-87 cells facilitated the cell cycle. We also examined change of cell cycle related proteins and found that TRIM29 regulated cyclin D1 and cyclin E expression. Cyclin D1 and cyclin E play a pivotal role during G1-S checkpoint and its overexpression correlates with bladder cancer aggressiveness [9-11]. Our results strongly suggested that TRIM29 promotes bladder cancer proliferation and cell cycle transition by upregulation of cyclin family proteins.
In the current study, we also found that overexpression of TRIM29 inhibited apoptosis and its depletion sensitized bladder cancer cell to cisplatin treatment. We observed that TRIM29 upregulated Bcl-2 expression. Bcl-2 functions as an anti-apoptotic protein in a number of cancers and its up-regulation results in prevention of cell death [12,13]. Thus, our results suggested that TRIM29 inhibited cancer cell apoptosis through upregulation of Bcl-2.
NF-κB is normally kept in an inactive state in the cytoplasm by binding to a group of inhibitory proteins. Upon receipt of a signal, its inhibitor is phosphorylated and proteolytically degraded and NF-κB is actively translocated to the nucleus, where it facilitates target-gene transcription. NF-κB signaling activation has been reported to promote cancer cell growth, angiogenesis, invasion, and eventually metastasis in many human tumors including bladder cancer [14,15]. Therefore, NF-κB along with its upstream and downstream network presents a rational target for therapeutic interventions. Our results showed an association between TRIM29 and NF-κB in bladder cancer cells. Both cyclin D1 and Bcl-2 were reported as downstream transcriptional targets of NF-κB [9,16,17]. To confirm their relationship, BAY 11-7082, NF-κB inhibitor, was used in treatment of TRIM29 transfected BIU-87 cells. The results showed that NF-κB inhibitor partially blocked the effect of TRIM29 on cyclin D1, Bcl-2, and cell proliferation, suggesting that TRIM29 regulates bladder cancer proliferation and apoptosis through, at least in part, NF-κB activation. PKC signaling acts as an upstream signaling molecule of NF-κB [18]. PKC was reported to increase phosphorylation of IκB and downstream targets including matrix metallopeptidase 9 in cancer cells [19]. Our results demonstrated that PKC inhibition blocked the role of TRIM29 in NF-κB activation, suggesting that the PKC–NF-κB axis plays a major role in TRIM29 induced proliferation and chemoresistance.
Conclusion
In conclusion, our study found that TRIM29 promoted cancer growth by facilitating cell cycle progression and inhibited cisplatin induced apoptosis in bladder cancer cells. TRIM29 also regulated expression of cyclin proteins and Bcl-2 through the PKC and NF-κB signaling pathways. The data implicated TRIM29 as a potential diagnostic and therapeutic target in bladder cancer.
 XML Download
XML Download