

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer is a common cancer which kills approximately one-third of the affected individuals in the developed world. It has been estimated that over 1 million people worldwide develop colorectal cancer annually [1]. Despite improved prognosis and treatment of colorectal cancer in recent years, no satisfactory therapy for colon cancer has been developed clinically because of associated tendency for metastasis. Initiation of metastasis requires invasion, which is enabled by the epithelial-mesenchymal transition (EMT). The EMT is a process whereby cells lose cell-cell adhesion and gain migratory and invasive properties. The EMT plays physiological roles in embryogenesis, wound healing, and tissue regeneration. In terms of pathology, it causes organ fibrosis; cancer development, progression, and metastasis; and chemoresistance. The EMT pathway has been considered a critical threshold that is crossed in the progression of most types of cancer involving migration [2]. During metastasis, cells from a primary cancer can invade adjacent tissue and enter the blood stream and lymphatic system, and circulate. Migrating cells can then attach to the endothelium of vessels and invade new sites and proliferate. Cancer metastasis is similar to the EMT–mesenchymal-epithelial transition (MET) process, and the EMT serves an important role in the metastasis of cancer [3].
The most common approach for investigating the EMT process is to study the expression of EMT biomarkers in human tissues or blood samples. Evidence of EMT in tissues or serum includes the expression of connective tissue markers such as vimentin, N-cadherin, fibronectin, and α-smooth muscle actin (SMA) and the downregulation or abolishment of epithelial markers such as E-cadherin and cytokeratin. Another characteristic of the EMT process is the upregulation of Snail family proteins (Snail and Slug), zinc-finger proteins (ZEB1, ZEB2), the HLH protein (Twist), and certain micro-RNAs.
Results of several studies have indicated that the EMT in metastatic cells depends upon various regulatory factors. Among those factors, E-cadherin, fibronectin, and Slug are useful prognostic indicators [4]. The phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin and mitogen-activated protein kinase (MAPK) signaling pathways are also involved in regulation of EMT [5]. Increased ARK phosphorylation has been implicated in the induction of the EMT and cell migration. In addition, the transcription factors Snail and Slug play essential roles in cell migration. Slug expression can be upregulated in the wound margins in in vitro, in vivo, and ex vivo settings. In Slug-null mice, re-epithelialization is reduced, compared with wild-type mice [4].
Cysteine-rich angiogenic inducer 61 (CYR61), a member of the CYR61/CTGF/NOV (CCN) protein family, consists of CYR61 (also known as CCN1 family member 1 [CCN1]), connective tissue growth factor (CTGF/CCN2), nephroblastoma-overexpressed (NOV/CCN3), and Wnt-induced secreted proteins 1, 2, and 3 (Wisp-1/CCN4, Wisp-2/CCN5, and Wisp-3/CCN6, respectively). These CCN proteins are involved in multiple functional pathways including mitogenesis, cellular adhesion, migration, cell survival, differentiation, angiogenesis, and wound healing [6]. Numerous studies have focused on the roles of CCN proteins in cancer, particularly those of CYR61 and CCN1. CYR61 may serve essential roles as either an oncogene or a tumor suppressor, depending on the cancer cell type. CYR61 also induces angiogenesis, which supplies oxygen and nutrients to tumors during growth. CYR61 can also play a role in the proliferation, invasion, survival, and metastasis of cancer cells [7].
Clinically, CYR61 expression is related to the prognosis of prostate cancer or breast cancer [8], although little is known about the role of CYR61 in colorectal cancer. Jeong et al. [9] measured CYR61 expression in 251 specimens from patients with colorectal cancer; in 6 cell lines (HT29, colo205, Lovo, HCT116, SW480, and SW620); and in 20 pairs of normal vs. colorectal cancer tissues.
Dexamethasone is an anti-inflammatory agent that blocks nuclear factor κB–induced cytokine transcription, resulting in inhibited cytokine production and macrophage activation [10]. In a murine model, corticosteroid administration prior to treatment with a chemotherapeutic agent resulted in reduced hematologic toxicity [11]. Dexamethasone is widely used as an effective anti-emetic in combination with chemotherapy, although it can potentially induce perioperative immunosuppression or promote tumor proliferation or metastasis [12]. However, these potential concerns were not manifested in a prospective human clinical trial with cancer patients. In a phase II randomized trial in patients with lung cancer who were treated with gemcitabine and carboplatin, pretreatment with dexamethasone prior to chemotherapy showed favorable effects, with reduced hematologic toxicity in patients. No significant difference in the overall survival was observed between the dexamethasone group and nondexamethasone group [13].
With many solid cancers (including colorectal cancer), dexamethasone is widely used to reduce the toxicity of chemotherapy. However, only a few clinical trials have been conducted to evaluate the impact of dexamethasone treatment on the survival of colon cancer patients. According to the results from a randomized, controlled trial conducted by Singh et al. [14] preoperative dexamethasone is associated with a higher rate of distant metastases in patients with colon cancer who undergo a colectomy.
Understanding the molecular mechanism of colorectal cancer progression is necessary to exploring various approaches for improved therapy. Here, we investigated the molecular mechanism whereby dexamethasone may promote human colorectal cancer cell migration and its relationship with CYR61 by co-treatment of HCT116 cells with dexamethasone with transforming growth factor β1 (TGF-β1), a cytokine that potentially promotes cellular migration.
Materials and Methods
1. Reagents
Human polyclonal anti-rabbit CYR61 antibody and human recombinant CYR61 were purchased from Santa Cruz Biotechnology (Dallas, TX). Recombinant TGF-β1 was purchased from Cell Signaling Technology (Danvers, MA). Dexamethasone was purchased from Sigma Aldrich (St. Louis, MO) and dissolved in distilled water to the indicated concentrations.
2. Cell culture
The HCT116 human colorectal cancer cell line was cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) and 1% penicillin/streptomycin (PAA Laboratories, Piscataway, NJ). Cells were incubated in a humidified atmosphere at 37°C in 5% CO2.
3. MTT assays
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was used in cell viability assays, as described in our previous study [15]. HCT116 cells were seeded in 96-well plates (200 μL/well, 3×104 cells/mL). Subsequently, the cells were treated with dexamethasone at various concentrations for 48 hours. MTT solution (10 μL, 5 mg/mL; Amresco, Solon, OH) was added to each well followed by incubation for 37°C for 4 hours. Subsequently, the medium was gently removed and replaced with 150 μL of dimethyl sulfoxide (DMSO) followed by incubation for 30 minutes with shaking to dissolve the precipitate. The samples were then analyzed by measuring the absorbance of 570 nm in a spectrophotometer.
4. Scratch wound healing assays
Cells were seeded at 50%-60% confluency in culture dishes for 24 hours before initiating wound healing scratch assays. A scratch was made in the cell monolayer by drawing a sterile p-200 pipette tip across the surface of the culture dish. After the scratch was made, the culture medium was supplemented with 10 ng/mL TGF-β1, 10 μM dexamethasone, or a combination of both. DMSO treatment was used as a vehicle control. At 0- and 48-hour post-treatment, photographs of the cell monolayer were acquired at 4× magnification using an Olympus IX70 microscope (Tokyo, Japan), equipped with a digital camera. The dimensions of scratches were measured using ImageJ software (National Institute of Health, Bethesda, MD), and the difference between the initial and final widths of the scratches was calculated.
5. Western blot analysis
Cells were treated with 10 ng/mL TGF-β1, 10 μM dexamethasone, or a combination of both for 48 hours. The cells were harvested by scraping and lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1.0% [v/v] NP-40, 0.5% [w/v] sodium deoxycholate, 1.0 mM ethylenediaminetetraacetic acid, 0.1% [w/v] sodium dodecyl sulfate [SDS], and 0.01% [w/v] sodium azide at a pH of 7.4). Protein concentrations were determined using the BCA Kit (Thermo Scientific, Grand Island, NY). Extracts were analyzed by SDS–polyacrylamide gel electrophoresis, followed by immunoblotting with appropriate antibodies. AKT (9272), phosphorylated AKT (9271S), ERK (4695), Slug (9585S), and glyceraldehyde 3-phosphate dehydrogenase (2118) were obtained from Cell Signaling Techonology. E-cadherin (610181) was purchased from BD Science Transduction (San Jose, CA), pERK (sc-7383), collagen I (sc-25974), collagen III (sc-28888), and fibronectin (sc-9068) were purchased from Santa Cruz Biotechnology. MMP1 (444209) was purchased from Calbiochem (Hessen, Darmstadt, Germany). We used antimouse (PI-2000, Vector Labs Inc., Burlingame, CA), anti-rabbit (PI-1000, Vector Labs Inc.), and anti-goat (AP-107P, Millipore, Billerica, MA) secondary antibodies. The blots were analyzed using ImageJ software. The relative changes in the ratios of expression of target proteins in treated cells to that in control cells were determined.
6. Reverse-transcriptase polymerase chain reaction experiments
Cells were seeded in a 60-mm culture dish and treated with 10 ng/mL TGF-β1, 10 μM dexamethasone, or a combination of both for 48 hours. RNA from treated cells was extracted using Trizol reagent (Invitrogen, Grand Island, NY). Next, 2 μg of total RNA were used for cDNA synthesis with the Reverse Transcriptase Kit (Promega, Madison, WI). The resulting cDNA was used for reverse-transcriptase polymerase chain reaction (RT-PCR) analysis with the G-Taq Kit (Cosmo Genetech, Seoul, Korea), according to the manufacturer’s instructions.
RT-PCR was performed using appropriate gene-specific forward and reverse primers, which were selected using the Blast Primer tool. The results were analyzed using the ImageJ software. The relative change in the ratio of the target protein to the DMSO control was determined.
7. Matrix metalloproteinases zymography
Matrix metalloproteinase (MMP) zymography was performed according to a standard procedure, with the modification that 8% SDS gels (Amresco) containing gelatin (0.01 mg/mL) were used. The SDS in the gels was removed by incubating the gels twice (30 minutes each) in 200 mL of 2.5% Triton X-100 at 4°C. Thereafter, the gel slabs were incubated at 37°C overnight in an incubation buffer (50mM Tris-HCl [pH 8.0], 5 mM CaCl2, 0.02% NaN3). The gels were then fixed and stained by immersion for 1 hour in 0.05% Coomassie Blue R-250 (Amresco). Molecular weight-marker proteins were visible as stained bands against the lighter blue stained gelatin background. Gelatinase activity was detected as clear zones (negative staining) against the blue background.
8. Transwell invasion assays
The Transwell invasion assay was performed using a Transwell plate (Corning, New York, NY). HCT116 cells were harvested by trypsinization, and 7×104 cells/well in 100 μL of medium was seeded into insert wells. The bottom well contained RPMI medium with 10% FBS as a chemoattractant. After 24 hours, the medium in the insert wells was replaced by 100 μL of serum-free medium containing different concentrations of dexamethasone and TGF-β1. Invasive cells in the receiving wells were collected by trypsinization and counted using a hemocytometer.
9. Statistics
Results are presented as the mean±standard deviation (SD). Group comparisons were performed with one-way analysis of variance and Student’s t test using SPSS ver. 12.0 (SPSS Inc., Chicago, IL). p < 0.05 was considered to indicate statistical significance. All experiments were repeated in at least three independent experiments.
Results
1. Dexamethasone inhibits TGF-β1–induced migration, but not colorectal cancer cell proliferation
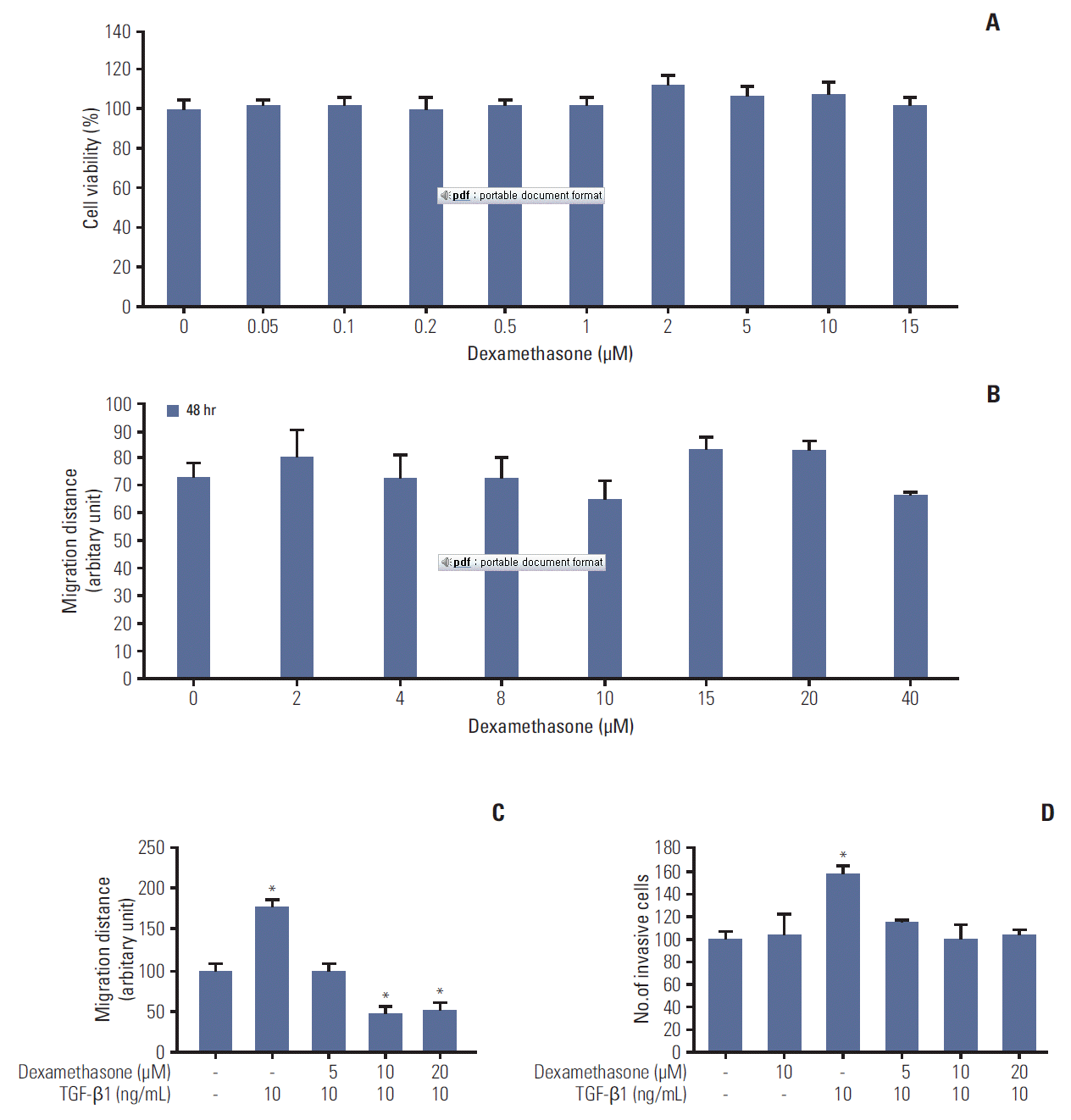
To examine the beneficial effects of dexamethasone in a human colorectal cancer cell line (HCT116), we first studied cell viability and migration in MTT assays and scratchwound assays, respectively. We observed no significant differences in cell viability in dexamethasone dose-response curves (Fig. 1A). Notably, the migration data indicated that treatment with dexamethasone resulted in significantly decreased cell migration at 10 μM after 24 hours, compared to untreated control cells (Fig. 1B).
TGF-β can induce EMT and the migration of various cell types [16]. Thus, we tested whether TGF-β1 can induce the migration and invasion of HCT116 cells. TGF-β1 (10 ng/mL) significantly augmented cell migration, whereas migration was significantly inhibited in cells co-treated with 10 ng/mL TGF-β1 and either 10 or 20 μM dexamethasone (Fig. 1C). To confirm this inhibitory effect of dexamethasone, we studied cell invasion in transwell assays, finding that dexamethasone inhibited TGF-β1–induced cell invasion (Fig. 1D). Collectively, these findings indicated that dexamethasone inhibited TGF-β1–dependent HCT116 cell migration, but not cell viability.
2. Dexamethasone inhibits the EMT process
Because dexamethasone inhibited TGF-β1–induced cell migration, we hypothesized that dexamethasone may also block TGF-β1–induced EMT. Thus, we tested whether dexamethasone treatment affects the expression of important EMT markers such as E-cadherin, vimentin, or Slug. At the transcriptional level, RT-PCR results indicated that although TGF-β1 induced elevated expression of Slug and vimentin, these levels were slightly, but not significantly, decreased in cells co-treated with dexamethasone. However, the TGF-β1–dependent decrease in E-cadherin was inhibited by co-treatment with dexamethasone (Fig. 2A). In addition, immunoblotting analysis showed that vimentin and Slug expression were similarly modified at the protein level by TGF-β1 and/or dexamethasone treatment (Fig. 2B).
Degradation of the extracellular matrix (ECM) by MMP proteins is required for tumor cells to undergo the EMT process. We found that MMP7 and MMP1 were slightly downregulated by TGF-β1 treatment, whereas treatment with dexamethasone resulted in upregulation of these metalloproteinases (Fig. 2C and D). In contrast, collagen type I and fibronectin, which are important ECM proteins involved in cell migration, were augmented by TGF-β1 treatment, while treatment with dexamethasone alone or in combination with TGF-β1 resulted in slightly decreased expression levels (Fig. 2C and D). In addition, gelatin zymography data showed that the levels of gelatinase activity from MMP2 and MMP9 were not changed significantly in treated cells compared to control cells (Fig. 2E). These data suggested that dexamethasone treatment inhibited the TGF-β1–induced EMT process, but was not directly related to ECM protein regulation.
3. Critical roles of the AKT and ERK signaling pathways in dexamethasone-inhibited migration
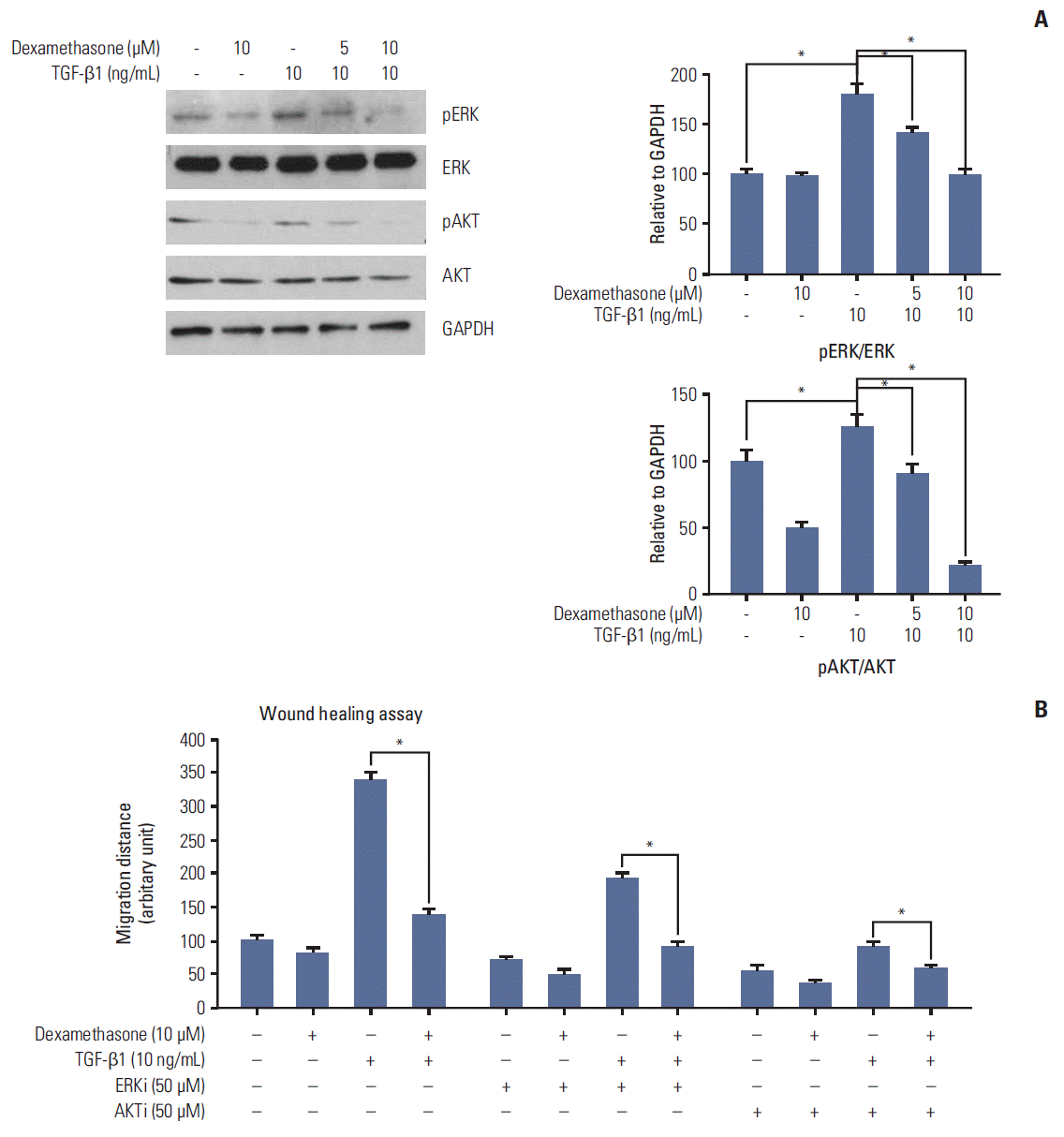
Activation of MAPK family members (ERKs) and the PI3K/AKT pathway is important for regulation of cell migration [17]. Therefore, we investigated the importance of the expression and phosphorylation of ERKs and AKT in cell migration by treating HCT116 cells with TGF-βwith or without dexamethasone co-treatment at differing concentrations. We found that TGF-β1 treatment induced AKT and ERK phosphorylation, which was inhibited in a dose-dependent manner by co-treatment with dexamethasone (Fig. 3A). Treatment with dexamethasone alone resulted in slightly decreased ERK phosphorylation and significantly decreased AKT phosphorylation. These data suggested that the inhibitory effect of dexamethasone on migration may involve AKT and ERK phosphorylation.
To confirm these findings, we investigated HCT116 cell migration following treatment with TGF-β1 treatment and/or dexamethasone in the presence or absence of the AKT inhibitor LY49002 (50 μM) or the ERK inhibitor PD98059 (50 μM). The inhibitors caused markedly decreased TGF-β1–dependent cell migration compared to control cells (Fig. 3B). Taken together, these findings demonstrated that TGF-β1 and dexamethasone regulated HCT116 cell migration via the ERK and AKT pathways.
4. CYR61-dependent regulation of migration and EMT via the AKT and ERK signaling pathways
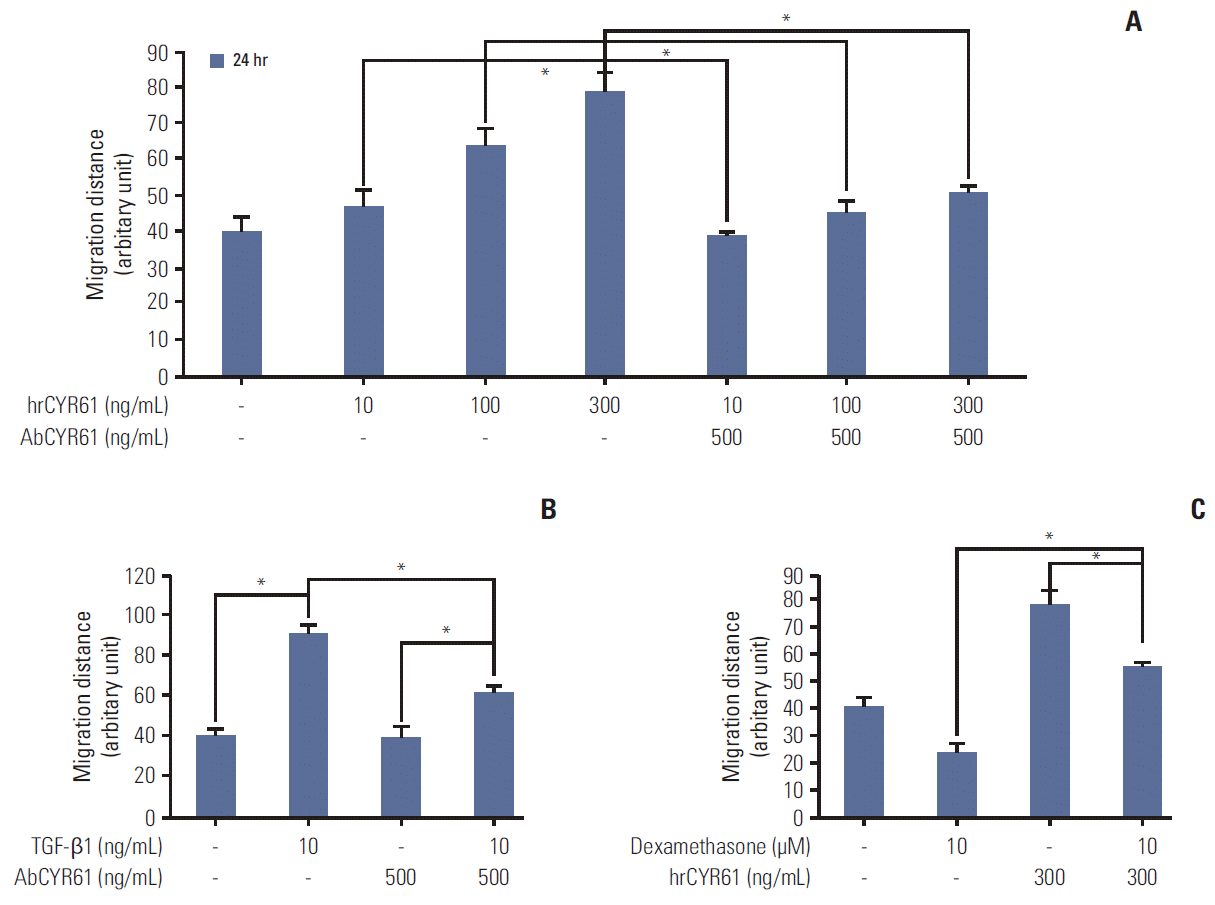
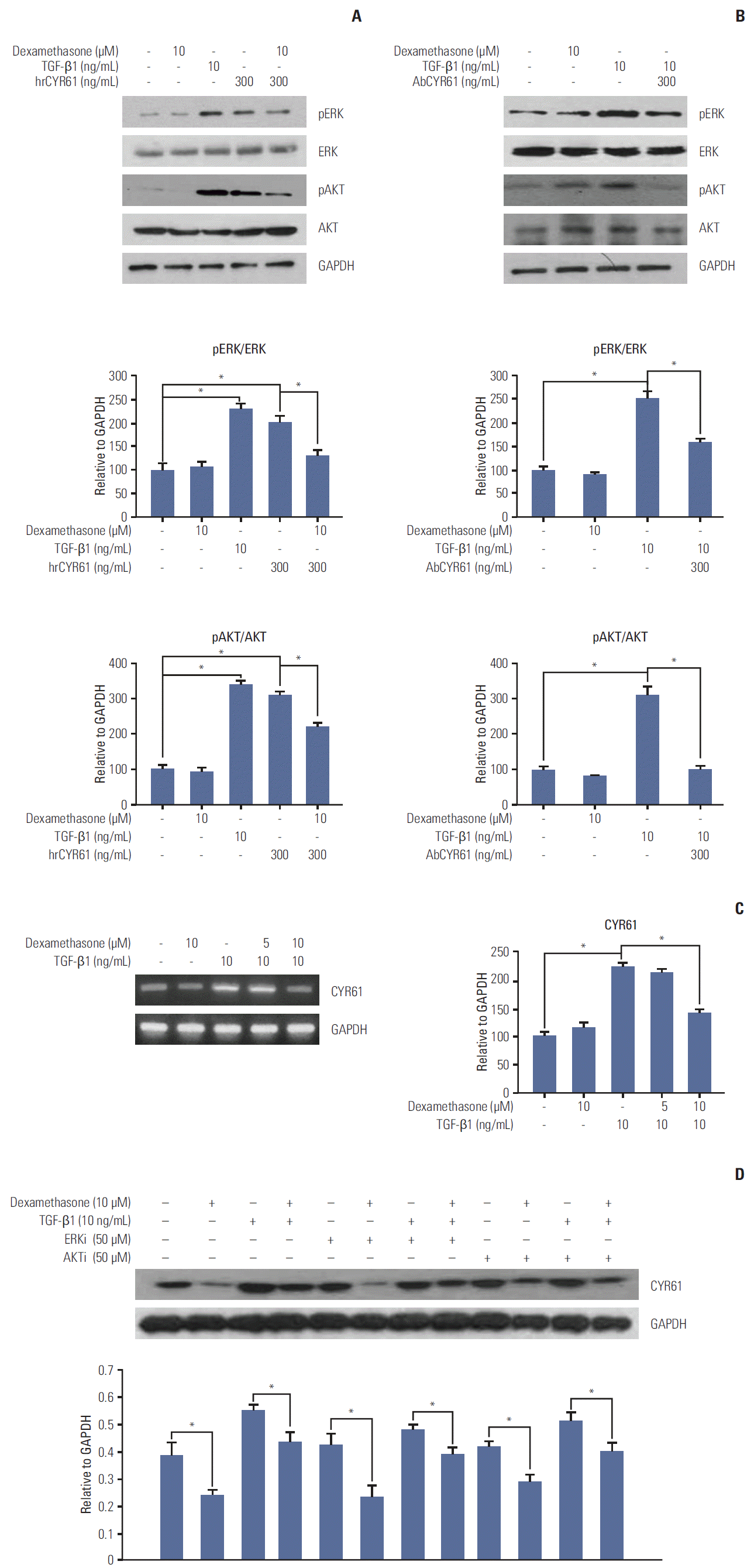
The findings described above demonstrated that TGF-β1 and dexamethasone can regulate cell migration via ERK and AKT phosphorylation. Thus, we hypothesized that the inhibition of migration observed by dexamethasone treatment in HCT116 cells may involve the CYR61 pathway. To determine the role of CYR61 in cell migration, HCT116 cells were treated with human recombinant CYR61 (hrCYR61), with or without an anti-CYR61 antibody (AbCYR61). Results from migration scratch assays indicated that hrCYR61 caused significantly increased cell migration in a dose-dependent manner, an effect that was abolished by AbCYR61 (Fig. 4A). TGF-β1–induced migration was also alleviated by treatment with AbCYR61 (Fig. 4B), whereas hrCYR61 counteracted dexamethasone-inhibited migration (Fig. 4C). Next, we examined the question of whether CYR61 participates in the ERK and AKT pathway by treatment of HCT116 cells with TGF-β1 alone, or by co-treatment with dexamethasone. As shown in Fig. 5A, ERK and AKT phosphorylation, which was downregulated by dexamethasone, was elevated by hrCYR61. Similarly, TGF-β1–induced ERK and AKT phosphorylation was reduced by AbCYR61 treatment (Fig. 5B). These results demonstrated that the CYR61 protein can regulate ERK and AKT phosphorylation.
To confirm that dexamethasone or TGF-β1 can affect cell migration through CYR61 modulation, we examined CYR61 expression following TGF-β1 and/or dexamethasone treatment. In agreement with the migration results described above, CYR61 mRNA and protein expression was increased by TGF-β1 treatment and abolished by co-treatment with dexamethasone (Fig. 5D). In addition, co-treatment with dexamethasone, TGF-β1, and either the ERK inhibitor PD98059 (50 μM) or the AKT inhibitor LY49002 (50 μM) showed that the effects of TGF-β1 and dexamethasone on CYR61 expression were not significantly changed by treatment with the ERK and AKT inhibitors, compared to control cells (Fig. 5D). These findings indicate that the CYR61 protein may serve as an upstream regulator of the ERK and AKT pathways during cell migration.
Discussion
CYR61, a secreted, ECM-associated signaling protein of the CCN family, is capable of regulating a broad range of cellular activities, including cell adhesion, migration, proliferation, differentiation, the EMT process, and apoptosis in several types of cancer cells [18,19]. TGF-β1 can upregulate CYR61 expression, leading to enhanced osteosarcoma cell migration [20]. CYR61 expression is modulated by extracellular acidification and PI3K/AKT signaling in prostate carcinoma PC-3 cells [21], and CYR61 signaling may be critical for EMT, stemness, and pancreatic carcinogenesis [19].
Glucocorticoids such as dexamethasone are widely used in combination with anti-cancer agents to reduce their adverse side effects. The benefits of glucocorticoids have been associated with their capacity to reduce tumor-associated edema. Recent findings have demonstrated that glucocorticoids operate through various mechanisms to alleviate the symptoms of chemotherapy in treatment of several types of cancer, including head and neck, breast, and cervical cancer. In head and neck cancer, dexamethasone treatment inhibits production of vascular endothelial growth factor via signal transducer and activator of transcription 3 suppression, leading to reduced cancer cell migration [22]. Dexamethasone may also be used in combination with other chemotherapeutics to improve their anti-cancer effects. For example, in combination with 1-alpha, 25-dihydroxyvitamin D [2], dexamethasone activated ERK/AKT signaling to transfer the signals to the nucleus and inhibit the cell cycle process, meaning that it enhanced the cell cycle arrest and might elevate cell apoptosis [23]. Despite these insights, the molecular mechanism whereby dexamethasone influences cancer cell migration remains unclear.
The data presented in this study demonstrated that dexamethasone treatment can significantly inhibit TGF-β1–induced migration and invasion of HCT116 colorectal cancer cells. However, dexamethasone treatment did not influence cell proliferation. Importantly, we observed that TGF-β1 promoted the EMT process, which was inhibited by dexamethasone. E-cadherin downregulation by TGF-β1 was reversed by co-treatment with dexamethasone (Fig. 2A and B). Downregulation of the cell junction protein E-cadherin appears to involve regulation by Wnt or TGF-β via the inducible activation of kinase pathways that modulate the activities of GTPases, SMADs, PI3Ks, MAPKs, and β-catenin, or the activation of transcription factors, including LEF-1, Snail, Slug, and Scatter, ultimately leading to E-cadherin repression [2]. Increased expression levels of Slug and vimentin can serve as markers for EMT [2]. In the current study, TGF-β1 was used to stimulate migration by inducing EMT (Figs. 1 and 2) in order to examine the effects of dexamethasone.
Results from several reports indicate that dexamethasone affects cell proliferation in cancer cells [21]. In contrast, our data showed that while dexamethasone reduced TGF-β1–induced migration, dexamethasone treatment alone did significantly affect migration. There was evidence that dexamethasone can inhibit EMT and promote MET. Dexamethasone has been shown to cause changes in cell morphology, expression of epithelial and mesenchymal cell markers (such as E-cadherin, ZO-1, and α-SMA), and cell migration [24]. In agreement, our results showed that dexamethasone treatment affected cell migration, as well as the expression of the epithelial cell marker E-cadherin and the transcription factor Slug (Fig. 3). However, dexamethasone treatment did not affect the EMT process. The expression of MMPs and collagen I was not significantly influenced by dexamethasone treatment.
We hypothesize that molecular signaling pathways modulate the effects of dexamethasone treatment. The MAPK and PI3K/AKT signaling pathways are associated with cell migration [17]. It has been previously demonstrated that glucocorticoids can affect MAPK and PI3K/AKT signaling activation. The beneficial effects of dexamethasone on TGF-β1–induced EMT occur by the amelioration of ERK and p38 MAPK phosphorylation. Similarly, we found that co-treatment with dexamethasone inhibited ERK and AKT phosphorylation, compared to TGF treatment alone. These data suggested that dexamethasone inhibited TGF-β1–induced EMT, which was confirmed by treatment with AKT and ERK inhibitors (Fig. 4).
Treatment with hrCYR61 significantly induced cell migration, a finding that was confirmed by showing that AbCYR61 treatment inhibited cell migration. In prostate carcinoma PC-3 cells, CYR61 protein production was modulated by extracellular acidification and PI3K/A signaling activation [21]. In osteosarcoma tumor and lung cancer cells, the CYR61 pathway was regulated by AKT and ERK phosphorylation [25]. In agreement, we found that hrCYR61 treatment induced AKT and ERK phosphorylation in human colorectal cells. In addition, ERK and AKT phosphorylation inhibitors did not affect CYR61 expression (Fig. 5D), although CYR61 expression was significantly reduced by TGF-β1/dexamethasone co-treatment compared with TGF-β1 treatment alone.
Conclusion
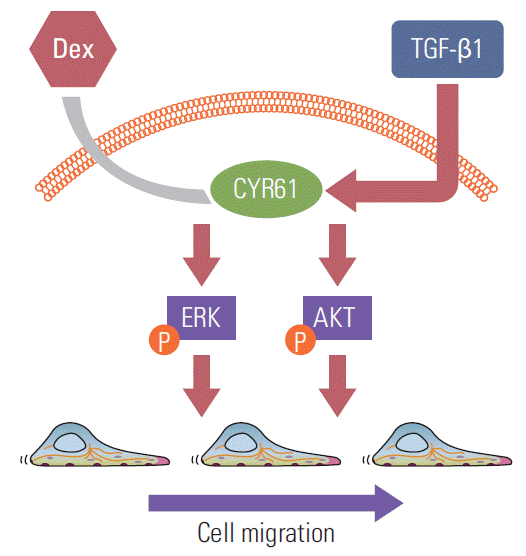
Taken together, our data are suggestive of a model whereby dexamethasone downregulates CYR61 expression, leading to decreased AKT and ERK phosphorylation and the reversal of TGF-β1–induced cell migration (Fig. 6).

 XML Download
XML Download