

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC) is a disease of epigenetic as well as genetic aberrancies. These genetic and epigenetic aberrances are not uniformly present in all patients as in a classic model of colon cancer evolution (adenoma-carcinoma sequence) [1]. Recent studies of colorectal carcinogenesis show that CRC is a heterogeneous disease arising from multiple pathways [2]. Knowledge of key molecular findings of each different pathway is not only helpful in explaining clinical outcomes but may also lead to more efficacious and individualized therapies.
The KRAS mutation is a validated biomarker of response to anti-epidermal growth factor receptor (EGFR) antibodies (cetuximab and panitumumab) [3,4]. In prospective randomized trials, the tumor mutation status of codons 12 and 13 of the KRAS gene was predictive for the activity of cetuximab combined with FOLFOX (oxaliplatin/leucovorin/5-fluorouracil) or FOLFIRI (irinotecan/leucovorin/5-fluorouracil) [5]. Therefore, performance of KRAS mutation analysis is mandatory before making treatment decisions. Regarding the prognostic role of KRAS gene, a prior international study found that KRAS mutations generally confer a worse prognosis [6]. However, conflicting results have been reported from analysis of recent large prospective trials [7].
CRCs can also be grouped according to epigenetic alterations, such as DNA methylation status. CpG island methylator phenotype (CIMP) is a distinct group with an increased frequency of aberrant promoter hypermethylation at specific loci. The “classic” CIMP markers (p16, MINT1, MINT2, MINT31, and hMLH1), which were initially suggested by Toyota et al. [8], defined two groups of CRC. In the study, CIMP positive CRCs had more frequent KRAS but fewer TP53 mutations than CIMP negative CRCs. The close association between CIMP and KRAS mutations as well as BRAF mutations was further reported in subsequent studies with classic markers [9,10]. The unique dependency on RAS/RAF pathway in CIMP CRCs might be predictive of anti-EGFR treatment.
The INK4a/ARF/INK4b locus (also known as CDKN2A and CDKN2B) on chromosome 9p21 encodes three genes (ARF, p15INK4b, and p16INK4a) [11]. Among them, the p16 gene encodes a G1 cyclin-dependent kinase (CDK) inhibitor that binds to and inactivates CDK4/6. Expression of p16 inhibits CDK4/6 mediated phosphorylation of retinoblastoma and results in G1 arrest in tumor cells [12]. The cell cycle arrest mediated by p16 upregulation is thought to be an important barrier to RAS activated oncogenic stress in colonic epithelial cells, termed oncogene-induced senescence [13]. In CRCs, inactivation of p16 is preferentially mediated by promoter hypermethylation of the p16 gene, one of the classic panels of CIMP [8,12]. In previous studies, alteration of p16, either by promoter hypermethylation or loss of expression, was associated with poor prognosis in patients with CRC [14-16]. In addition, a preclinical study reported that p16 gene hypermethylation was associated with decreased response to irinotecan in colon cancer cell lines and a demethylating agent, 5-azacytidine, enhanced the anti-cancer effect [17].
In this study, we retrospectively evaluated the ability of CIMP status and p16 gene hypermethylation status to predict best objective response (BOR), time to progression (TTP), and overall survival (OS) in CRC patients treated with cetuximab-FOLFIRI (E-FOLFIRI) chemotherapy.
Materials and Methods
1. Patient characteristics
We included 49 patients with metastatic or recurrent CRC who were treated with 5-fluorouracil, leucovorin, irinotecan, and cetuximab (E-FOLFIRI) as first-line (22 patients) or second-line (27 patients) therapy. All patients were treated at Severance Hospital of Yonsei University from January 2005 to January 2011. Clinical data were obtained from electronic medical records of Severance Hospital and survival data were retrieved from the tumor registry at Yonsei Cancer Center. Exclusion criteria included co-existing malignancies (except for non-melanoma skin cancer or in situ cervical cancer), cancer other than adenocarcinoma, and lack of availability of formalin-fixed paraffin-embedded (FFPE) tumor tissue. This study was approved by the institutional review board (IRB) at Yonsei University Severance Hospital (Seoul, Korea).
2. Treatment and efficacy assessment
E-FOLFIRI chemotherapy consisted of weekly cetuximab (initial dose 400 mg/m2 intravenously [IV] over 2 hours, and 250 mg/m2 IV weekly, over 1 hour, thereafter) and biweekly FOLFIRI (irinotecan 180 mg/m2 IV on day 1, leucovorin 200 mg/m2 IV on day 1, 5-fluorouracil [5-FU] 400 mg/m2 IV bolus on day 1 followed by 2,400 mg/m2 IV over 46 hours, every 2 weeks). FOLFIRI was administered after 1 hour of cetuximab infusion. Treatment was continued until disease progression or unacceptable toxicity. Tumor response was evaluated after four cycles (every 8 weeks) by computed tomography scan and classified according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria ver. 1.1.
3. DNA methylation and KRAS mutation analysis
Genomic DNA from FFPE tissue was extracted using QIAamp DNA FFPE tissue kit (Qiagen, Valencia, CA). DNA extracted from FFPE tissue was used to evaluate the methylation status of six CpG islands (MINT1, MINT2, MINT31, hMLH1, p16, and p14). Bisulfite treatment of DNA was performed before pyrosequencing using an EZ Methylation kit (Zymo Research, Orange, CA) according to the manufacturer’s protocol. Pyrosequencing was performed using the PyroMark Q24 instrument (Qiagen) according to the manufacturer’s protocol. The detailed protocol for pyrosequencing of DNA methylation has been described in previous reports [10]. The methylation status of CpG island markers was considered to be methylation positive using a threshold value of 15% (negative, methylation level < 15%; positive, methylation level ≥ 15%). A tumor was considered CIMP positive if two or more CIMP markers were methylation positive.
Mutation status of KRAS gene was also determined with pyrosequencing assays. Mutations of codons 12 and 13 were determined using the PyroMark Q24 instrument (Qiagen) [10].
4. Statistical analysis
Differences in BOR rates between the two groups (CIMP positive vs. negative; p16 methylated vs. unmethylated) were analyzed using a two-sided Fisher exact test. The TTP and the OS were estimated using the Kaplan-Meier method and compared using the log-rank test. A Cox proportional hazard model was used to examine the independent contribution of each variable that was considered to be a potential predictive variable for survival in univariate analysis. Regression model included variables that showed significant association with survival in univariate analysis and three variables of interest: age, sex, and CIMP status. A value of p < 0.05 was considered significant, and all resulting p-values were two sided. Analyses were performed using SPSS for Windows ver. 18.0 (SPSS Inc., Chicago, IL).
Results
1. Patient characteristics
A total of 49 CRC patients treated with E-FOLFIRI were selected based on sample availability. The baseline characteristics of the patients are summarized in Table 1. p16 gene hypermethylation was observed in 14 of 49 tumors (28.6%). There was no significant difference in patient age, Eastern Cooperative Oncology Group (ECOG) performance status, histology, location of primary tumor, and prior chemotherapy between p16 methylated and p16 unmethylated tumors. Metastatic disease and recurrent disease at the time of treatment accounted for 77.1% and 22.9% of patients without p16 methylation and 35.7% and 64.3% in p16 methylation, respectively (p=0.006). Mutations in KRAS codons 12 or 13 were significantly more common in p16-methylated tumors (42.9%) than in p16-unmethylated tumors (8.6%, p=0.01). p16 methylation showed significant association with CIMP (p=0.002).
2. Tumor response and survival according to p16 methylation and CIMP
Among 49 evaluable patients, 44 patients had one or more measurable lesions and five patients had no measurable disease. The BOR (complete response+partial response) rate by RECIST was 68.8% in patients with unmethylated p16 (22/32; 95% confidence interval [CI], 51.5 to 84.8) and 33.3% (4/12; 95% CI, 9.1 to 62.5) in patients with methylated p16 (Fisher exact test, p=0.045) (Table 2). Compared with CIMP negative, patients with CIMP positive tumors had a lower response rate (40% [6/15; 95% CI, 16.7 to 69.2] vs. 69% [20/29; 95% CI, 50.0 to 85.2]), but this was not statistically significant (Fisher exact test, p=0.064).
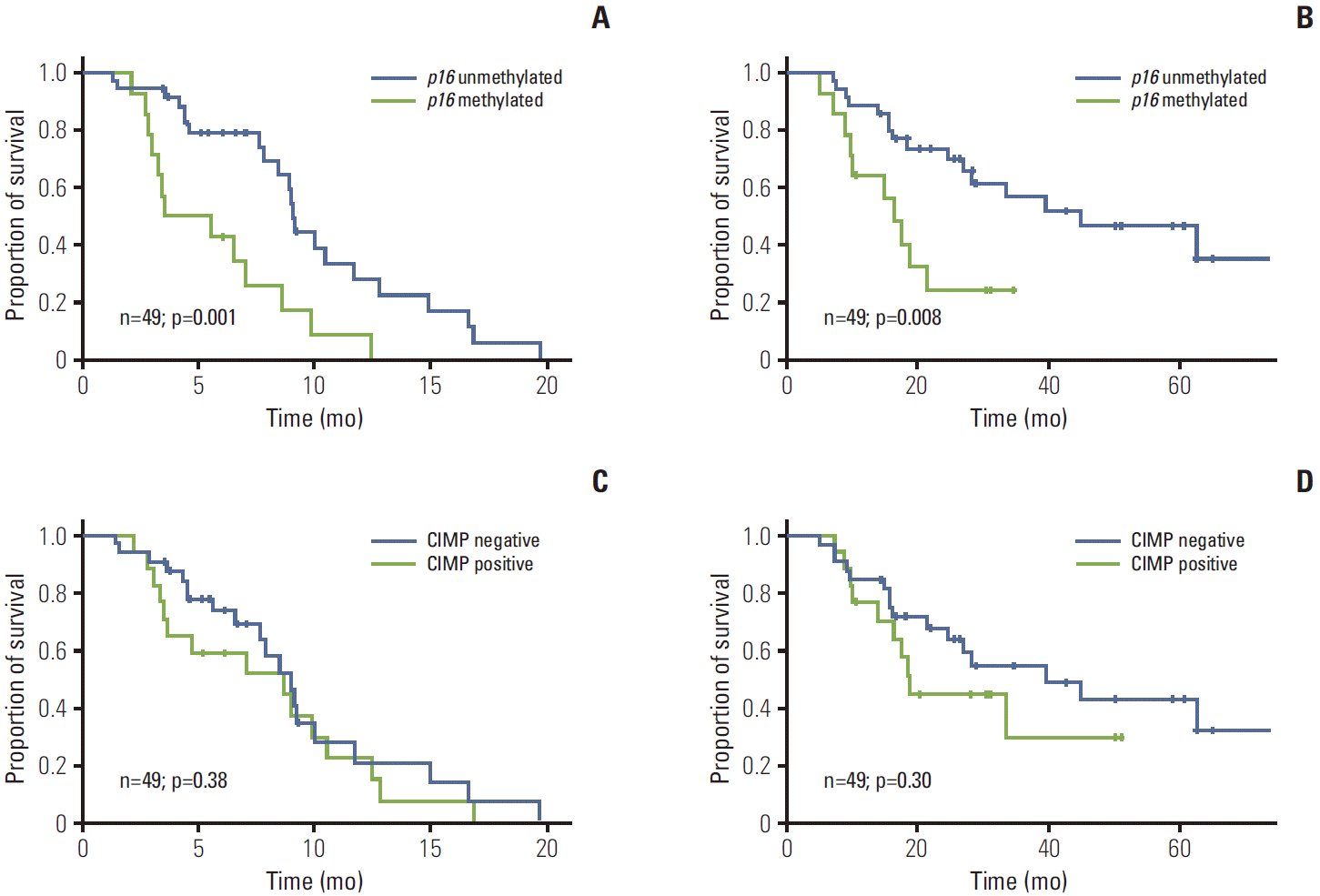
Median TTP was 9.0 months (95% CI, 8.8 to 9.3) in the unmethylated p16 group and 3.5 months (range, 0.0 to 7.4 months) in the methylated p16 group (hazard ratio [HR], 3.15; 95% CI, 1.52 to 6.54; log-rank, p=0.001) (Fig. 1A). In the unmethylated p16 group, 12 of 35 patients (34.3%) with unresectable disease were converted to resectable status and underwent surgery, while, on the contrary, none of the methylated p16 patients was able to undergo surgery. With a median follow-up period of 21.4 months (range, 5.1 to 74.3 months), median OS was 44.9 months (95% CI, 18.1 to 71.7) in the unmethylated p16 group and 16.4 months (range, 12.0 to 20.7 months) in the methylated p16 group (HR, 2.95; 95% CI, 1.28 to 6.81; log-rank, p=0.008) (Fig. 1B). In our cohort, however, CIMP status was neither a significant predictor of TTP nor of OS (p=0.38 and p=0.30, respectively) (Fig. 1C and D). After correcting for significant predictive factors for TTP by multivariate analysis, p16 methylation was associated with poorer TTP (p=0.027; HR, 2.97; 95% CI, 1.14 to 7.74), but not with OS (p=0.071) (Table 3).
3. Subgroup analysis stratified by KRAS mutation and p16 methylation
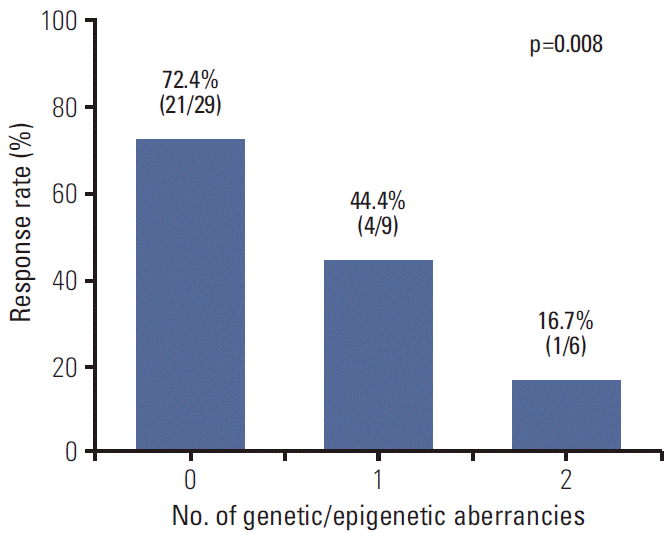
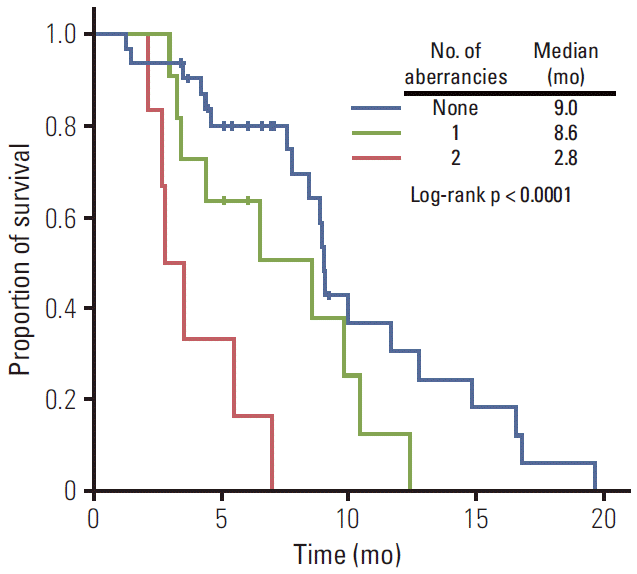
For a more detailed analysis of response to E-FOLFIRI treatment, we classified patients according to the number of genetic (KRAS mutation) and epigenetic aberrancies (p16 methylation). Response rate significantly decreased as the number of aberrancies increased (Fig. 2). The univariate odds ratio of response between the no aberrancy group and 2 aberrancy group was 13.12 (95% CI, 1.32 to 130.42; p=0.028). Median TTPs of no aberrancy group, 1 aberrancy group, and 2 aberrancy group were 9.0 months, 8.6 months, and 2.8 months, respectively (log-rank, p < 0.0001). The Kaplan-Meier curve for TTP of each group showed a trend toward worse outcome (Fig. 3).
Discussion
The primary aim of this study was to explore the use of methylation status in predicting outcome after E-FOLFIRI therapy for metastatic CRC. We have shown that epigenetic alteration of p16 is independently associated with poorer outcome in these patients. In addition, patients with both KRAS mutation and p16 methylation had remarkably poor prognosis despite E-FOLFIRI treatment. These findings suggest that epigenetic alteration, in addition to genetic aberrancies such as KRAS mutation, may be helpful in defining which patients are likely to benefit from systemic treatment in CRC patients.
It should be noted that CRC patients with both p16 methylation and KRAS mutation have shorter survival than those with either one alone or no aberrancies. The role of p16 as a tumor suppressor is well established in a variety of human cancers [11,12]. It has been postulated that p16 plays a significant role in the signal transduction of the RAS/RAF pathway and acts as a barrier to oncogenic stimulation. In CRCs, Esteller et al. [14] reported shorter survival of CRC patients with KRAS mutation and/or p16 methylation after surgical resection compared to those with neither of those alterations. In a variety of tumors with RAS/RAF activation, such as melanoma, pancreatic cancer, and malignant astrocytoma, frequent concomitant loss of p16 has been observed and conferred resistance to chemotherapeutic agents [18]. Recently, in a preclinical study that utilized CRC cell lines, p16 demethylation by 5-azacytidine enhanced cytotoxicity of irinotecan treatment [17]. In addition, inhibition of CDK4/6 with PD0332991, a novel selective CDK 4/6 inhibitor, instead of p16 could be another potential targeted agent. It has already demonstrated antitumor activity in a human CRC xenograft model and showed significant clinical activity in hormone receptor positive breast cancer patients [19]. Therefore, combining demethylating agents or PD0332991 with chemotherapeutic agents or RAS/RAF pathway inhibitor might be a rational approach to overcome resistance and improve outcomes in patients with this aggressive tumor type.
CIMP subtype was not a powerful predictor of clinical outcome in our study. CIMP subtype did not show significant association with TTP and OS, despite a trend toward lower response rate in CIMP positive. Previous studies regarding molecular characteristics of CIMP subtype have shown that CIMP positive tumors are characterized by a high frequency of KRAS and BRAF mutations [9,10,20]. On the basis of this molecular characteristic of CIMP, it has been suggested that CIMP status might be a possible predictive marker for anti-EGFR therapy. In addition, patients with CIMP positive tumors were associated with mismatch repair deficiency and tend to show a worse response to 5-FU chemotherapy [21]. A study by Ogino et al. [22] reported that CIMP status predicted treatment response and survival in microsatellite stable CRC patients after 5-FU and irinotecan based combination chemotherapy. For the subtype specific personalized therapeutic approach in CRC, further studies are needed to elucidate the exact mechanism and overcome the inherent chemoresistance of CIMP subtype.
Biomarker identification in cancer tissue is a crucial step in development of personalized treatment. The clinical impact and usefulness of biomarkers are dependent on their stability and reproducibility. In contrast to the relatively low stability of protein or RNA based biomarkers, the advantage of DNA methylation marker is stability over time. DNA methylation in gene promoters remains stable over years in cancer cell lines and xenograft models [23]. A recent study demonstrated that gene expression of unmethylated promoters is quite variable but gene expression is stably repressed when the promoters are methylated [24]. Moreover, the reversibility of DNA methylation by hypomethylating agents suggests that they are not only surrogate markers of response but also potential targets for treatment. This point may be addressed by an ongoing clinical trial (Clinical Trial.gov identifier No. NCT01105377).
We note that our study had several limitations. First, study results must be interpreted with caution because of the small number of patients who were analyzed without a control arm. Therefore, these results should be validated in a larger setting in prospective cohorts. Second, BRAF mutation analysis was not performed in our study. As KRAS and BRAF mutations are mutually exclusive [20], poor survival in patients with methylated p16 and KRAS mutation is not affected by BRAF status. In addition, recent studies suggest that the less common mutation of KRAS (in exon 3 or 4) and NRAS predicted a lack of response in patients who received anti-EGFR antibody [25]. Last, we utilized “classic” CIMP panels because various groups using this panel have found consistent results in CRC patients [8,10,20]. However, the absence of a consensus panel defining the CIMP subgroup is a hurdle to be overcome with future investigations.
Conclusion
In conclusion, we found that hypermethylation of p16 was predictive of clinical outcome in CRC patients treated with E-FOLFIRI, irrespective of KRAS mutation status. Although CIMP status was not predictive of E-FOLFIRI response, there was a trend toward poor responsiveness. These findings should be validated in further prospective trials for individualized cancer treatment.
 XML Download
XML Download