

 PDF
PDF Citation
Citation Print
Print


Introduction
The mammalian forkhead transcription factors of the O class (FOXOs) have four members: FOXO1, FOXO3, FOXO4, and FOXO6. FOXO1 and FOXO3 are expressed in nearly all tissues [1]. When FOXOs are phosphorylated, they are exported from the nucleus and do not exhibit transcriptional activity [2]. In recent years, FOXOs have caught much research attention for their broad roles in physiological process, including cell cycle arrest, apoptosis, angiogenesis, stress resistance, energy metabolism, and stem cell differentiation [3]. Dysregulation of FOXOs results in disease, including cancer. Inactivation of the FOXO1 occurs in many types of human cancer, including endometrial cancer, breast cancer, ovarian cancer, prostate cancer, rhabdomyosarcoma, hemangiosarcoma and gastric cancer (GC) [1].
Sirtuins are NAD+-dependent deacetylase enzymes in mammals and consist of SIRT1-7. They are localized in different cellular compartments and are capable of many catalytic activities [4]. Among them, SIRT1 is the most thoroughly studied and is expressed in the brain, heart, liver, pancreas, skeletal muscle, spleen, and adipose tissues [5]. SIRT1 regulates physiological events that include oxidative stress, metabolism, cellular proliferation, and genomic stability via deacetylation of histone and non-histone proteins [5]. Furthermore, SIRT1 targets many transcription factors, including p53, FOXO, E2F1 and nuclear factor kB, and consequently is involved in diverse functions [6].
Currently GC is the second most frequent cause of cancerrelated death [7]. The incidence of GC continues to rise despite effective treatment that increases the survival of GC patients. However, the prognosis remains poor [8]. Although several pathways are suggested as a mechanism of GC angiogenesis, the underlying molecular mechanism remains largely unknown. We previously reported that FOXO1 inactivation was commonly found in surgically obtained GC specimens, associated with angiogenesis-related molecules [1].
SIRT1 controls the nuclear shuttling and transcriptional activity of forkhead transcription factors and regulates FOXO activity either positively or negatively depending on the target gene or cell type [9]. Though the association between FOXO1 and SIRT1 was gradually elucidated in various disease conditions, there has not been any study of that association in GC, let alone angiogenesis.
Here, we extended our previous study and further investigated the direct role of FOXO1 in GC angiogenesis. FOXO1 expression was suppressed by RNA interference and the effects of FOXO1 silencing on the GC growth and angiogenesis were assessed in cell culture experiments and a nude mouse model of subcutaneous xenografts. In addition, we assessed the relationship between FOXO1 and SIRT1 using human GC tissue array and xenograft tumors.
Materials and Methods
1. Cell cultures
Five well-characterized human GC cell lines (SNU-638, SNU-601, SNU-216, SNU-484, and SNU-668) were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI 1640 medium (Life Technologies, Grand Island, NY) containing 10% fetal bovine serum, incubated in 5% CO2, and either 20% (normoxic) or 1% (hypoxic) O2 atmosphere at 37°C.
2. Western blot analysis
Western blot analysis was performed as we described previously [10]. Cell lysates in sodium dodecyl sulfate (SDS) lysis buffer (125 mM Tris-HCl [pH 6.8], 4% SDS, 0.004% bromophenol blue, and 20% glycerol) were separated on 10% SDS–polyacrylamide gel and electrophoretically transferred to PVDF membranes (Millipore Co., Billerica, MA) blocked with 5% non-fat dry milk in phosphate buffered saline– Tween-20 (0.1%, vol/vol) for 1 hour. The membranes were then incubated with a primary antibody against FOXO1 (1:1,000, C29H4, Cell Signaling Technology, Beverly, MA), hypoxia inducible factor-1α (HIF-1α; 1:500, H1α67, BD Biosciences, San Jose, CA), vascular endothelial growth factor (VEGF; 1:1,000, C1, Santa Cruz Biotechnology, Santa Cruz, CA), SIRT1 (1:1,000, H300, Santa Cruz Biotechnology), or β-actin (1:1,000, C4, Santa Cruz Biotechnology). Horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000, Santa Cruz Biotechnology) or anti-mouse IgG (1:2,000, Santa Cruz Biotechnology) was used as a secondary antibody. Enhanced chemiluminescence (Amersham, Arlington Heights, IL) was used to detect the immunoreactive proteins. Equal protein loading was confirmed by β-actin.
3. Lentivirus-mediated short hairpin RNA silencing of FOXO1
Lentiviral particles containing FOXO1 short hairpin RNA (shRNA) or non-targeting shRNA, control shRNA, were purchased (Sigma, St. Louis, MO). The sequence of FOXO1 shRNA was 5′-CCGGGCCTGTTAT CAATCTGCTAAACTC GAGTTTAGCAGATTGAT AACAGGCTTTTTG-3′. The control shRNA particles contain four base pair mismatches within the short hairpin sequence to any known human or mouse gene. Viral infection was introduced by incubating GC cells in the culture medium containing lentiviral particles for 12 hours in the presence of 5 μg/mL Polybrene (Santa Cruz Biotechnology). Pooled puromycin (2 μg/mL)-resistant cells were used for further analysis.
4. Luciferase reporter assay
To determine FOXO1 nuclear DNA-binding activity, luciferase reporter assay was used as previously described with slight modifications [10,11]. Two oligonucleotides (GCAAAACAAACTTATTTTGAAGCAAAACAAACT TATTTTGA AGCAAAACA AACT TATTTTGAA and TTCA AAATAAGTTTGTTTTGCTTCAAAATAAGTTTGTTTTG TT CAAAATAAGTTTGTTTTGC) were annealed and ligated into the pGL4.27-Promoter vector (Promega, Madison, WI) to create 3XIRS-luciferase (Cosmo GENETECH, Seoul, Korea). This construct has three tandem repeats of a FOXO1 binding element, the insulin-responsive sequence (IRS), inserted upstream of the luciferase reporter gene. It is widely used as an indicator of FOXO1 transcriptional activity. GC cells were cotransfected transiently with 0.4 μg 3XIRS-luciferase vector and 0.4 μg pSV-β-galactosidase vector (Promega), an internal control, using Lipofectamine Plus (Life Technologies). Twenty-four hours after transfection, assays for luciferase and β-galactosidase were carried out using a Dual-Luciferase Reporter Assay System (Promega). FOXO1 luciferase activity was measured on an AutoLumat LB 9505c luminometer (Berthold Analytical Instruments, Nashua, Germany) and was normalized by β-galactosidase activity.
5. Assessment of cell viability
SNU-638 (2.5×104 cells) and SNU-601 cells (2.5×104 cells) were seeded into each well of 24-well plates and were allowed to grow for 0 to 24 hours. Cell numbers were measured indirectly using the method reported by Kim et al. [12]. Cells were stained with 0.2% crystal violet aqueous solution in 20% methanol for 10 minutes, dissolved in 10% SDS, transferred into 96-well plates, and the absorbance was measured at 570 nm using an enzyme-linked immunosorbent assay reader (Bio-Rad, Hercules, CA).
6. Mouse xenograft model
All animal procedures were performed in accordance with the procedures described in the Seoul National University Laboratory Animal Maintenance Manual (approval No. SNU-120412-5). Six-week-old male nude mice (BALB/cSlcn/n) were purchased from SLC Inc. (Hamamatsu, Shizuoka, Japan) and maintained under specific pathogen-free conditions. For subcutaneous implantation, tumors were established by injecting SNU-638 GC cells expressing either control shRNA or FOXO1 shRNA at the density of 5×106 cells in 100 μL of Matrigel (R&D Systems, Minneapolis, MN) subcutaneously into the left flank of each mouse. Mice were divided into two groups by FOXO1 expression in GC cells: control shRNA or FOXO1 shRNA. Tumor volumes were measured every two days using a caliper, and were calculated by the equation V (mm3)=(length×width×height)× (π/6). Animals were sacrificed 48 days after cell implantation, and tumor xenografts were removed and prepared for immunohistochemistry or Western blot analysis.
7. Patients and tissue array methods
Four hundred and seventy-one surgically resected human GC specimens were obtained from the Department of Pathology, Seoul National University College of Medicine from January 2 to December 29, 2006. Nine paraffin tissue array blocks were prepared as previously described [13]. This protocol was reviewed and approved by the Institutional Review Board of the Seoul National University (approval No. C-1006-035-320).
8. Tumor histology and immunohistochemistry
Tissue specimens from clinical GC samples and xenograft tumors derived from GC cells were fixed with 10% neutralbuffered formalin, and 4-μm paraffin sections were prepared. After rehydration, sections were stained with hematoxylin and eosin for histologic assessment, or were immunostained after antigen retrieval using a Bond-max automated immunostainer (Leica Microsystems, Newcastle upon Tyne, UK). The primary antibodies used were against FOXO1 (1:40, C29H4, Cell Signaling Technology), phospho-FOXO1Ser256 (pFOXO1; 1:60, Cell Signaling Technology), CD31 (1:100, M20, Santa Cruz Biotechnology), HIF-1α (1:50, provided by Dr. Jong-Wan Park at Seoul National University), VEGF (1:200, C1, Santa Cruz Biotechnology), and SIRT1 (1:100, H300, Santa Cruz Biotechnology). Antibody binding was detected with the Bond Polymer Refine Detection Kit (Leica Microsystems). All immunostained sections were lightly counterstained with Mayer’s hematoxylin. Throughout the above analysis, negative controls were prepared by omitting the primary antibody. The results of immunostaining were evaluated by two pathologists (Y.K. and J.-S.P.), who were blinded to the origin of the samples. For statistical analysis, the results of immunostaining for proteins were considered positive if immunoreactivity was seen in ≥ 10% (cytoplasmic pFOXO1 and nuclear SIRT1) or ≥ 5% (nuclear HIF-1α) of the neoplastic cells.
9. Quantification of microvessel areas
Microvessel areas (MVAs) in subcutaneous xenograft tumors were determined by light microscopy/optical image analysis after immunostaining xenograft tumor sections with anti-CD31 as described previously [10]. The three most highly vascularized areas in areas of tumors near the tumor– normal tissue interface were selected. Photographs of CD31-immunopositive vessels in tumor sections were taken under a light microscope, and the cross-sectional areas of CD31-immunopositive structures (i.e., microvessel areas) were quantified by capturing images, converting them to grey scale, and analyzing CD31-stained areas using NIH Image Analysis software after setting one consistent intensity threshold for all slides. CD31-positive areas were expressed as pixels squared per high-power field and were measured for all tumors.
10. Statistical analysis
For cell culture and animal experiments, data were analyzed using GraphPad Prism software for Windows 7 (ver. 4, GraphPad Software, San Diego, CA). A two-tailed Student’s t test was used to rule out the role of chance in the results. For tissue array analysis, statistical analyses were conducted using SPSS ver. 19.0 (IBM Co., Chicago, IL), and the chi-squared test was used to determine the relationship between the expressions of pFOXO1Ser256, SIRT1, and HIF-1α. Results were expressed as mean value±standard deviation. Null hypotheses of no difference were rejected if p-values were less than 0.05.
Results
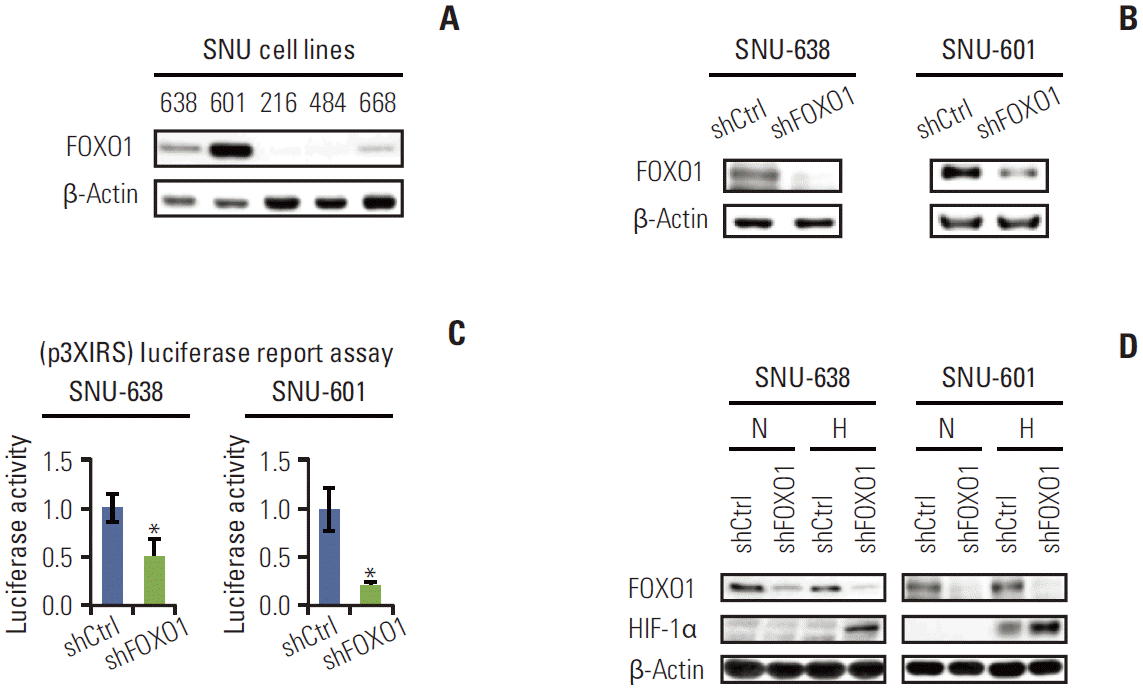
1. FOXO1 shRNA expression effectively inhibited FOXO1 expression and transcriptional activity in GC cell lines
To investigate the molecular effects of FOXO1 in GC, we initially examined the expression level of FOXO1 in GC cell lines. The protein contents of FOXO1 varied in five different cell lines (Fig. 1A). To observe the effect of FOXO1 silencing in the GC cell, SNU-638 and 601 cell lines with high levels of FOXO1 expression were selected, and FOXO1 expression was suppressed using RNA interference. Western blot analysis (Fig. 1B) and luciferase reporter assay (Fig. 1C) verified that transfection of FOXO1 shRNA effectively inhibited FOXO1 expression and its transcriptional activity in both cell lines.
2. FOXO1 silencing increased the protein expression of HIF-1α in GC cells under hypoxic conditions, but not under normoxic conditions
HIF-1α, which regulates many genes involved in adapting to a hypoxic environment, is important in promoting gastric tumor growth and angiogenesis [14]. Therefore, we performed cell culture experiments using GC cell lines SNU-638 and SNU-601 to examine the direct relationship between the expressions of FOXO1 and HIF-1α. GC cells expressing control shRNA or FOXO1 shRNA were cultured under normoxic or hypoxic conditions. By Western blot, FOXO1 down-regulation corresponded with increased HIF-1α protein expression under hypoxic conditions, but not under normoxic conditions, in both cell lines (Fig. 1D).
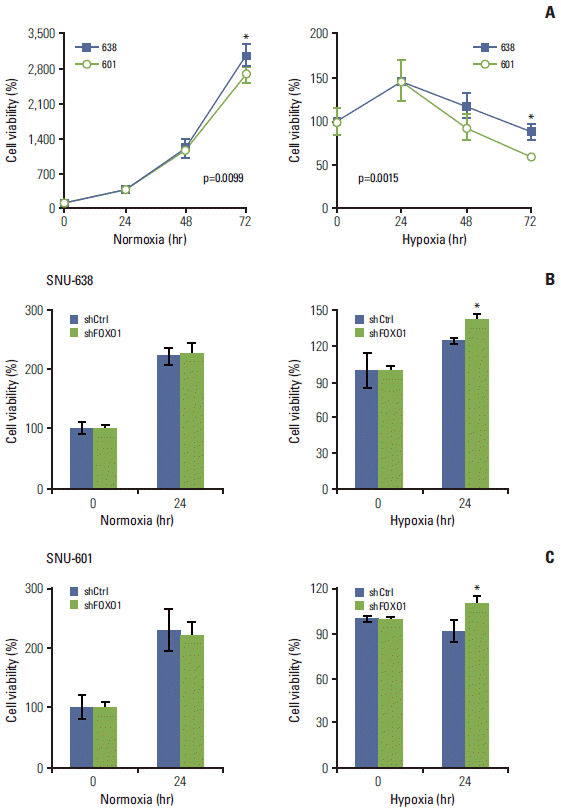
3. FOXO1 silencing increased GC cell viability under hypoxic conditions
Fig. 1A shows that FOXO1 expression was higher in SNU-601 cells than in SNU-638 cells. In contrast, we found that cell growth rate was higher in SNU-638 cells than in SNU-601 cells, which became more evident under hypoxic conditions (Fig. 2A). To confirm the growth-inhibitory role of FOXO1, we compared the viability of GC cell lines under normoxic and hypoxic conditions. At 24 hours of hypoxic exposure, in both GC cell lines FOXO1 knockdown increased cell viability under hypoxic conditions, but not under normoxic conditions (p < 0.05) (Fig. 2B and C). These data suggest that the effect of FOXO1 on GC cell growth needs a hypoxic environment.
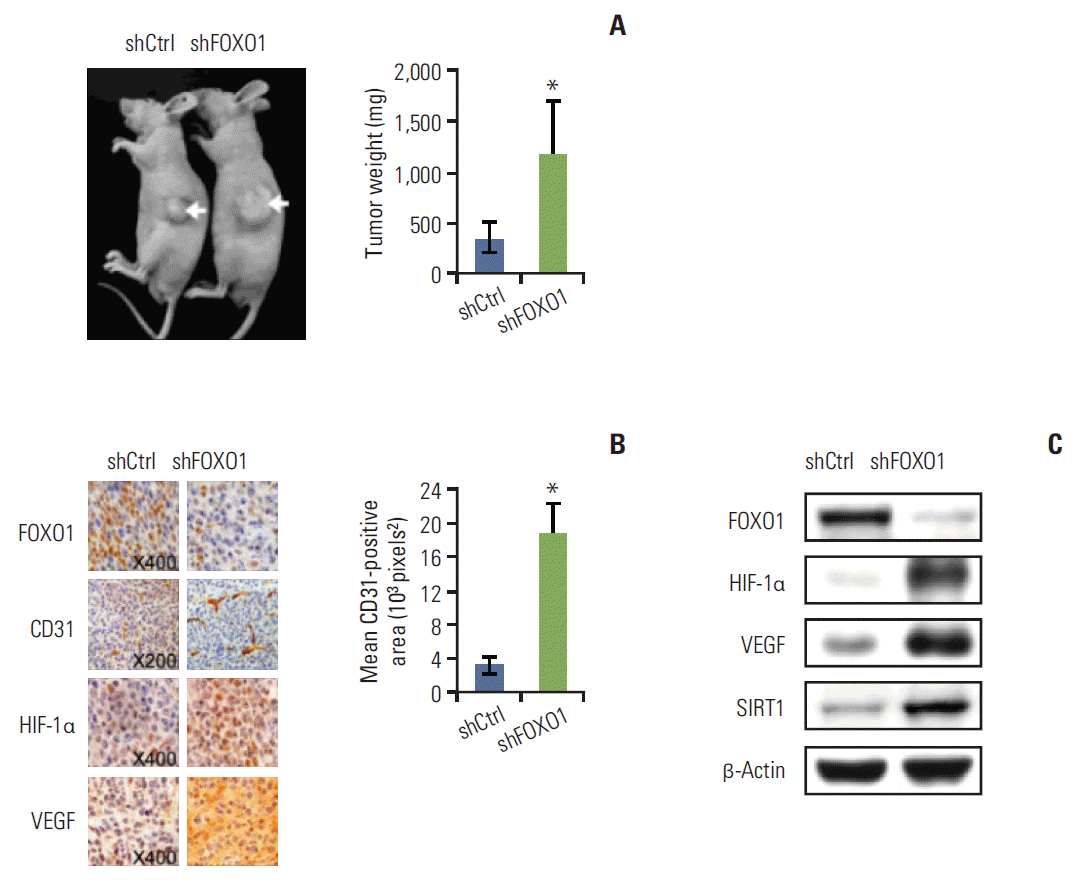
4. FOXO1 silencing increased tumor growth, MVA and the expression of HIF-1α, VEGF, and SIRT1 proteins in a nude mouse model of subcutaneous GC xenografts
Fig. 2A shows that SNU-638 cells grow faster than SNU-601 cells. Thus, to examine the role of FOXO1 in GC growth, we injected SNU-638 cells rather than SNU-601 cells into nude mice. Stable SNU-638 cells expressing non-targeting (control) or FOXO1-targeting shRNA were used. We found that tumors were successfully formed in all mice. The tumor weights were markedly higher in mice injected with FOXO1 shRNA expressing cells than those injected with control shRNA expressing cells (p < 0.05) (Fig. 3A).
Next, tumor tissues were removed and prepared for immunohistochemistry (Fig. 3B, left) and Western blot analysis (Fig. 3C). FOXO1 shRNA tumors showed much lower FOXO1 protein expression than the control shRNA tumors (Fig. 3B, top row), verifying the direct inhibitory role of FOXO1 in tumor growth. Angiogenesis in xenografted tumors was assessed using immunohistochemistry for CD31, a specific marker of vascular endothelial cells. CD31 immunoreactivity increased in FOXO1 shRNA tumors compared to control shRNA tumors (Fig. 3B, second row). Optical image analyses showed that the CD31-positive MVA was noticeably larger in FOXO1 shRNA tumors (mean, 18.56×103 pixels2) than in control shRNA tumors (mean, 3.25×103 pixels2) (Fig. 3B, right). Thus, FOXO1 silencing increased MVA in GC xenografts. In addition, FOXO1 shRNA increased the expression of HIF-1α and VEGF, both of which promote GC tumor growth [15], angiogenesis [15] and metastasis [16] (Fig. 3B, third and fourth rows; Fig. 3C, second and third rows). These results strongly suggest that FOXO1 functions to inhibit tumor growth through negative regulation of tumor angiogenesis mediated by the HIF-1α– VEGF pathway.
In addition, the association found between FOXO1 and SIRT1 was observed in xenografted tumors. Western blot analysis showed that FOXO1 shRNA-tumors showed higher SIRT1 expression than control shRNA-tumors (Fig. 3C).
5. FOXO1 shRNA knock-down in GC cells up-regulated the expression of SIRT1
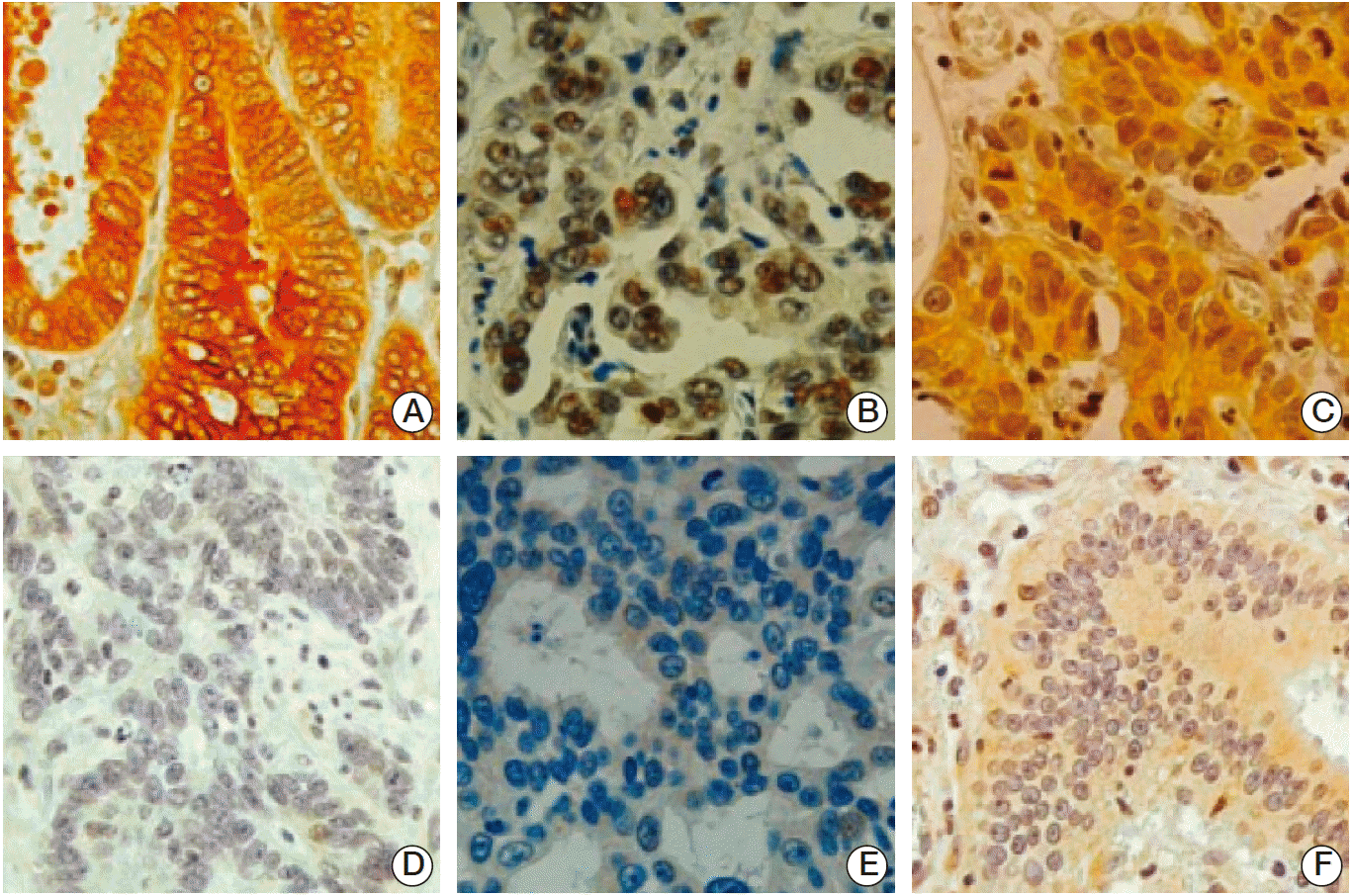
SIRT1 increases gastric tumor growth and angiogenesis [17]. Thus, we examined the relationship between the expressions of FOXO1 and SIRT1. In immunohistochemical tissue array analysis of human GC samples, pFOXO1 (Fig. 4A and D), SIRT1 (Fig. 4B and E), and HIF-1α (Fig. 4C and F) were expressed in both the nuclei and cytoplasm of tumor cells. Regarding pFOXO1 expression, cells showing distinct cytoplasmic staining, with or without the presence of nuclear staining, were considered to express the inactive form of FOXO1 constitutively. With SIRT1 and HIF-1α expressions, nuclear staining was considered to indicate activation of these two molecules. We analyzed the association between FOXO1 inactivation (pFOXO1 expression) and the activations of SIRT1 and HIF-1α in 471 human GC specimens. Table 1 shows that cytoplasmic pFOXO1 expression was found in 359 (77%), nuclear SIRT1 expression in 178 (38%) and nuclear HIF-1α expression in 108 of the 471 (23%) of human GC cases. This result shows positive associations between pFOXO1 and SIRT1 (p < 0.001) and HIF-1α (p < 0.001). Moreover, a positive association was found between SIRT1 and HIF-1α (p < 0.001).
Discussion
Though important tumorigenic processes such as cell proliferation or anti-apoptosis are often targeted in conventional anticancer treatments, tumor angiogenesis is considered the most important predictor of overall survival in GC [18]. Therefore, it is obvious that finding a potent therapeutic target should be preceded by a detailed understanding of the mechanisms underlying angiogenesis. However, the mechanisms underlying GC angiogenesis are far from understood. A major goal of this study was to confirm our previous hypothesis that FOXO1 has an anti-angiogenesis effect through inhibition on angiogenesis-related molecules.
Growing evidence shows that FOXOs are significant in angiogenesis. FOXOs are involved intimately in endothelial cell development and angiogenesis [19]. Several possible pathways might explain FOXOs in angiogenesis. In part, FOXOs interact with HIF-1 at multiple levels as hypoxia-induced FOXO-dependent Cited2 expression blocks HIF-1 at transcriptional level [20]. In our previous study, FOXO1 was negatively related to several angiogenesis-related molecules such as HIF-1α and VEGF in 272 surgical samples of GC [1]. In the present study, FOXO1 silencing in GC cell lines increased HIF-1α expression and cell viability under hypoxic conditions. Consistently, animal studies showed that FOXO1 shRNA-tumors grew faster and developed larger MVA as well as higher expressions of HIF-1α and VEGF than control shRNA-tumors. Thus, these data confirm the inhibitory role of FOXO1 in the tumor growth and angiogenesis in GC.
SIRT1 acts paradoxically in tumor angiogenesis, both inhibiting [6] and activating [21] angiogenesis. Regarding GC, an earlier study showed that VEGF silencing down-regulated the expression of SIRT1 as well as tumor growth in a nude mouse model of subcutaneous xenografts [22]. In the present study, a positive association was found between the nuclear expressions of SIRT1 and HIF-1α by immunohistochemical tissue array analysis of 471 human GC specimens, suggesting a positive regulatory role of SIRT1 in GC angiogenesis.
During oxidative stress, FOXOs translocate to the nucleus and interact with SIRT1, resulting in the deacetylation of FOXOs. FOXOs may be tightly modulated by SIRT1 during tumorigenesis [23,24]. Dependent upon the post-translational changes on FOXOs, SIRT1 can inhibit FOXO activity to protect cells from oxidative stress [23] or increase the activity of FOXOs to lead to gene activation [24]. The present study showed a positive association between the expression of inactive form of FOXO1 and nuclear expression of SIRT1 in human GC specimens. Furthermore, animal studies showed that FOXO1 silencing increased SIRT1 expression in GC xenografts. Thus, our data and those from a previous study [22] indicate that FOXO1 suppresses GC angiogenesis through HIF-1α/SIRT1 pathway. Although the relationships between FOXO1 and HIF-1α and that of SIRT1 and HIF-1α have been reported [1,6], the FOXO1-SIRT1 pathway that ties to regulation of angiogenesis in GC or any type of cancer has not been. FOXO1 may suppress tumors through different strategies depending on the tumor type. For the first time, we report that FOXO1 suppresses angiogenesis through the HIF-1α and VEGF and FOXO1-SIRT1 pathway.
The pursuit of understanding the mechanisms that regulate vascular growth in tumors will continue to expand and deliver new ideas for therapeutic exploration. Regulation of FOXO1-SIRT1 pathway in GC promises great potential for investigating the pathogenesis of GC and identifying a novel therapeutic target, as well as a useful biomarker.
Conclusion
In conclusion, this study provided an insight into the biological behavior of FOXO1 in GC angiogenesis. Since FOXO1 is functionally inactivated in a high percentage of GCs, inhibition of angiogenesis through restoration of FOXO1 expression and subsequent inactivation of HIF-1α, VEGF and/or SIRT1 may be an attractive approach for treatment of GC.
 XML Download
XML Download