

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC) has been a significant cause of morbidity worldwide [1]. Although patients diagnosed with early stage disease have a high cure rate, many are diagnosed later with a poor 5-year survival. The treatment of CRC has improved significantly over recent years with new generation chemotherapeutic agents and molecular targeted agents. Recent efforts to improve treatment have resulted in a new step toward individualized healthcare. Several studies have shown that a treatment of metastatic CRC with antibodies against epidermal growth factor receptor (EGFR) is effective only among patients with wild type Kirsten-ras (KRAS) carcinomas, whereas mutant type KRAS is predicted to have resistance to this treatment [2]. Some studies have indicated that there is a presence of KRAS mutation in lung cancer and that CRC correlates with poor prognosis [3,4]. Thus, determination of KRAS status is now recommended in patients with advanced CRC who are selected for EGFR targeted therapies. KRAS is a proto-oncogene, encoding a small 21-kD guanosine triphosphate/guanosine diphosphate binding protein involved in the regulation of cellular response to many extracellular stimuli [5]. Mutations within KRAS abrogating the GTPase activity and resulting in the activation of RAS/RAF signaling are found in 35% to 42% of CRCs and are thought to occur early in CRC carcinogenesis. There was a limited number of mutations in the KRAS gene, and more than 90% of these involve three codons: 12, 13, and 61 [6].
Most KRAS mutations have been identified from surgical or biopsy tissue. However, it is sometimes difficult to obtain tumor tissue from patients with inoperable CRC or tumor DNA from non-surgical tumor tissue, which have derived from endoscopic biopsy. Even in a prospectively conducted clinical trial, < 50% of patients had tumors that were available for mutation analysis [7]. Circulating DNA fragments carrying tumor specific sequence alterations (circulating tumor DNA) are found in the cell free fraction of blood, representing a variable and generally small fraction of the total circulating DNA [8,9]. Mutant circulating serum DNA has been studied in the serums of patients with various tumors including CRC [10,11]. Anker et al. [11] reported that KRAS mutation was detectable in the plasma of 86% (6 of 7) of CRC patients in whom KRAS mutation was present in the primary site. The serum sample can be obtained repeatedly and noninvasively from all CRC patients irrespective of patients’ characteristics. Thus, serum might be used as a sample for serial monitoring to changes of specific genetic mutation according to tumor progression.
In this single institute, prospective study, we analyzed 65 patients with advanced CRC for KRAS mutation in codon 12 and 13 by using a polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) assay for serum sample and genomic polymerase chain reaction (PCR)/direct sequence method for matched tumor tissue to identify the role of the serum sample as an alternative candidate for detection of KRASmutation. We also investigated the potential implication of KRAS mutation as predicting the outcomes in advanced CRC patients.
Materials and Methods
1. Patients
Patients were required to have a histologically proven metastatic or recurrent CRC, an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 1, and no previous chemotherapy excluding adjuvant therapy, between March 2008 and July 2011. As soon as they were diagnosed with metastatic or recurrent CRC, serum samples from these patients were obtained. Only patients treated at the Korea University Anam Hospital were included in this analysis. Clinicopathologic data were recorded in the registry of solid cancer of the oncology division. Clinicopathologic parameters recorded were as follows: age, sex, ECOG PS, disease status, operation, tumor location, sites of metastasis, laboratory finding at the start of chemotherapy, the status of KRAS mutation in tumor tissue, and chemotherapy regimen. All patients gave permission for the use of their serum and tumor tissue.
2. KRAS mutation analysis in tissue
DNA was extracted from five paraffin sections of 10-μm thickness containing a representative portion of the tumor tissue (Qiagen, Hilden, Germany). Fifty nanograms of DNA were amplified in a 20-μL reaction solution containing 10 μL of 2× concentrated HotStarTaq Master Mix (Qiagen), including polymerase chain reaction buffer, 3 mM MgCl2, 400 uM each of dNTP, and 0.3 μM each of the primer pairs (codon 12, 13, F: 5′-CGTCTGCAGTCAACTGGAAT, R: 5′-GAGAATGGTCCTGCACCAGTAA). Amplifications were performed using a 15-minute initial denaturation at 95°C, followed by 35 cycles of 30 seconds at 94°C, 30 seconds at 59°C, and 30 seconds at 72°C, and a 10-minute final extension at 72°C. The PCR products were then 2% gel-purified using the QIAgen gel extraction kit (Qiagen).
DNA sequencing was conducted as follows: first, digested mutated DNA was used as a template for the second PCR, in which the primer Ras 3 antisense (5'-GGATGGTCCTCCACCAGTAATATGGATATTA-3′) was used instead of the Ras 2 (antisense: 5′-TTATCTGTATCAAAGAATGGTCCTGCACCA-3′). The PCR was run under the same conditions as the first PCR for 32 cycles. Because of the nested antisense primer (Ras 3), the second PCR generated a fragment of 152 bp. This mutated DNA was excised from 3% agarose gels. Amplicons were then purified using the High Pure PCR Product Purification kit (Boehringer-Mannheim, Mannheim, Germany). Five nanograms of the purified amplicons were used for sequencing, which was performed with the Big Dye RR Terminator reaction (ABI, Weiterstadt, Germany). The product was run on a 5% polyacrylamide gel in an ABI 373A Sequencer (ABI), which was then analyzed for point mutations of the respective amplicons.
3. Blood sample collection and DNA extraction
Blood samples were collected at the time of diagnosis in metastatic or recurrent CRC. The volume of each blood sample was 10 mL. The serum was separated within 2 hours from collection and stored at –80°C until using. Serum DNA was extracted and purified by using a Qiamp Blood Kit (Qiagen). One column was used repeatedly until the whole sample had been processed. The resulting DNA was eluted in 50 μL of sterile bidistilled buffer. The concentration and purity of the extracted DNA were determined by a spectrophotometer. The extracted DNA was stored at –20°C until used.
4. PCR-RFLP assays
Mutations at KRAS codons 12 and 13 in the serum sample were analyzed by a highly sensitive PCR-RFLP: 300 ng of DNA was used as a template for the first PCR, which consisted of 50-μL volume containing Taq DNA polymerase (Takara, Kyoto, Japan), deoxynucleotide triphosphates (Takara), reaction buffer (Takara), and oligonucleiotide primers RAS1 (sense: 5′-ACTGAATATAAACTTGTGGTCCATGGAGCT-3′) and RAS2 (antisense: 5′-TTATCTGTATCAAAGAATGGTCCTGCACCA-3′). For amplication, a PCR machine (BioRad, Hercules, CA) was used. The PCR generated an amplicon of 166-bp length. Cycling conditions of the PCR were as follows: initial denaturation (5 minutes at 98°C), followed by 35 cycles of denaturation (10 seconds at 98°C), annealing (20 seconds at 53°C), and elongation (1 minute at 72°C). After the last cycle, a final extension (5 minutes at 72°C) was added; thereafter, samples were kept at 4°C. The RAS 1 (sense) primer (mismatch bases are underlined) had been designed to introduce a restriction site for BstXI and XcmI into the wild type amplicon. Because of this altered sequence, BstXI (5′-CCANNNNNNTGG-3′) restricted the resulting amplicon if the first two bases of codon 12 (underlined bases) were wild types. Similarly, XcmI (5′-CCANNNNNNNNNTGG-3′) cut the amplicon only if the first two bases of codon 13 (underlined bases) were wild types. For the restriction, 10 μL from PCR reaction were digested with five units of either BstXI or XcmI in the total volume of 20 μL. Twenty-two milliliters of the product was run on 5% polyacrylamide gel (Roth, Karlsruhe, Germany), stained by ethidium bromide for 1 minute, and analyzed under ultraviolet (UV) light using a video densitometer (Herolab, Wiesloch, Germany).
5. Statistical analysis
The chi-test or Fisher exact test was used to assess the association between KRAS mutation status and each of the clinicopathologic parameters. The relationship between KRAS mutations detected in the serum and tumor samples was evaluated by a correlation analysis (p-value and correlation index). A p-value of < 0.05 was considered statistically significant. Overall survival (OS) according to KRAS mutation status in the serum and tumor samples was estimated by the Kaplan-Meier method compared using the 2-sided log-rank test. The Cox proportional hazard modeling method was applied for a multivariate analysis of OS.
Results
1. Patients’ characteristics
Sixty-five patients were enrolled between March 2008 and July 2011. The median age of all patients was 62 years (range, 35 to 82 years) at diagnosis, and the male:female ratio was 1.7:1.0. The median ECOG PS was 1 (range, 0 to 1). Table 1 presents the baseline characteristics of all patients. The primary lesion of most patients was located in the left side colon. Twelve patients had recurrent disease and 41 of 65 received cytoreductive operation without curative intent. The majority of patients (73.8%) had more than 2 metastatic lesions and 44 patients revealed liver metastasis. KRAS mutations were detected in 40.0% of the tested blood sample and 47.7% of the tumor sample. All patients (n=65) received first-line chemotherapy. Eight of 65 were treated with irinotecan-based regimen and others with oxaliplatin-based regimen. Fifty-five of 65 had second-line chemotherapy. Of the ten patients without second-line chemotherapy, four did not experience the disease progression and six were not appropriate for receiving second-line chemotherapy.
2. Sensitivity and specificity of detection for KRAS mutation in serum DNA
In pairs of tumor and serum samples from 65 patients, the concordance of KRAS mutation between the tumor and serum samples was detected in 18 pairs. KRAS mutations were detected in the serum samples of 26 patients and in the tumor samples of 31 patients. KRAS mutation status was consistent in 44 of the 65 pairs (67.7%). There was a significant correlation between the mutation detected in the tumor sample and the mutation detected in the matched serum sample (p < 0.004; correlation index, 0.35) (Table 2).
3. Correlation between KRAS mutation status and survival
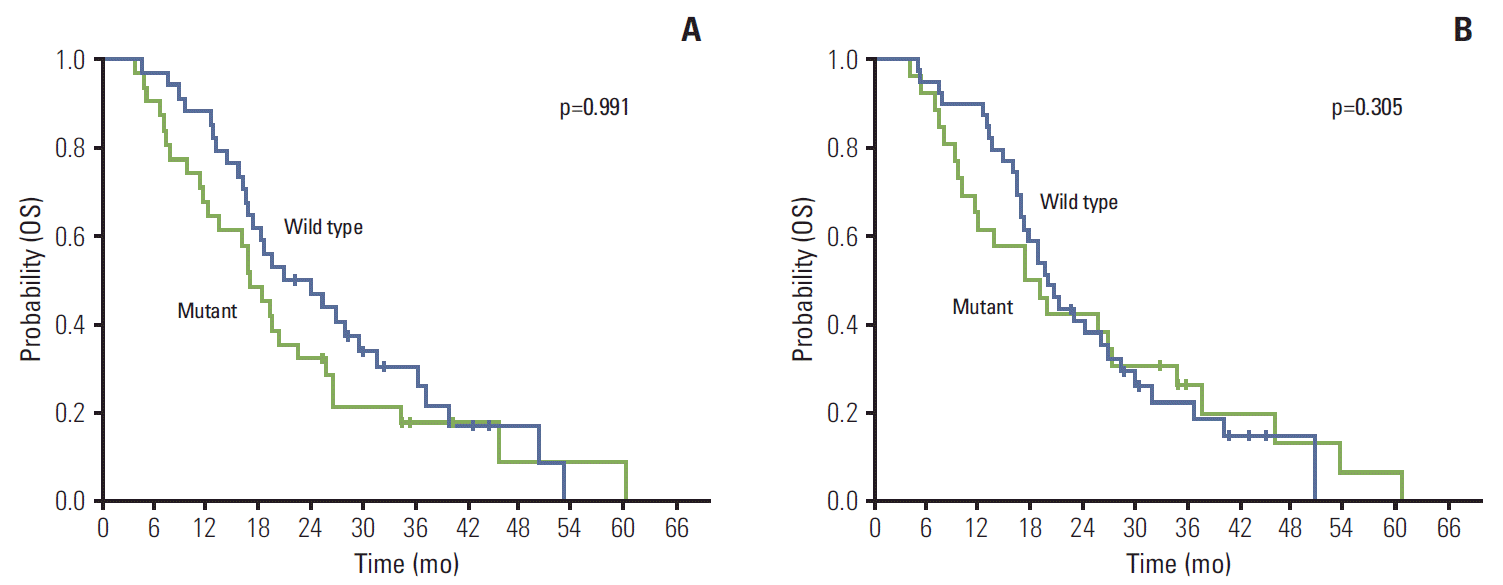
There were no patients’ characteristics having significant association with KRAS mutation status in both the tumor and serum samples (Table 3). There was no significant difference for OS in 65 patients according to KRAS mutation status in the serum samples (p=0.991) (Fig. 1A). This finding was identical on an analysis for KRAS mutation status in the tumor samples (p=0.305) (Fig. 1B). In analysis of 55 patients with two or more line chemotherapy, there was no difference for OS according to KRAS mutation status in the serum and tumor samples.
4. KRAS mutation in both serum and tumor samples as a prognostic factor
Univariate analysis showed that decreased OS was significantly associated with liver metastasis, no debulking operation, and over more normal level of cancer antigen 19-9 (≥ 37 IU/mL). In a multivariate analysis, liver metastasis (hazard ratio [HR], 1.940; 95% confidence interval [CI], 1.039 to 3.624; p=0.038), and no debulking operation (HR, 2.023; 95% CI, 1.123 to 3.642; p=0.019) were also significantly associated with decreased OS. KRAS mutation in both the serum and tumor samples did not have the impact as a prognostic marker for survival (Table 4).
Discussion
KRAS mutation has been used widely as the predictive marker for EGFR-targeted monoclonal antibodies in patients with metastatic or recurrent CRC [12,13]. Although there exist a controversy, KRASmutation has been as a good prognostic marker independent on the use of EGFR targeted therapies in some studies [3,14]. Thus, what we know the status of KRAS mutation is very important to select patients who might have more benefit from EGFR targeted therapies and to predict the course of the disease. Our findings revealed that the serum sample might alternatively be used when it is difficult to get the tumor tissue for an analysis of the KRAS mutation status in patients with advanced CRC. There was a high correlation between mutations detected in the serum sample and mutations detected in the matched tumor sample (correlation index, 0.352; p < 0.004). KRAS mutation status was consistent in 44 of the 65 pairs (67.7%). Our result showed a discordance with the outcomes of Anker et al. [11]. This inconsistency among studies is considered due to the heterogeneity of genetic abnormalities in tumors and the difference of race.
It is important to understand the KRAS mutation status to identify patients who might benefit from EGFR monoclonal antibody therapies and to make decisions in clinical practice. Several studies have indicated that the presence of KRAS mutation in CRC correlates with poor prognosis [3,15]. Besides, it is known that the changes in the gene status of the tumor might have a correlation with the passage of time [16,17]. This change in the gene status can be a consequence of the natural evolution of the tumor or as a result from the therapy. Knowing the serial genetic changes of the tumor are essential to realizing the personalized healthcare. Through monitoring genetic changes, we can avoid continuing ineffective therapies, prevent unnecessary side effect, and determine the benefit of new therapeutics in accordance to the current tumor genetics. However, it is sometimes difficult to obtain the tumor samples needed for genetic analysis. Moreover, for an analysis of the genetic changes, it will be more difficult to obtain serial tumor specimens in accordance to the disease progression or treatment. Advances in sequencing technologies have enabled rapid identification of genomic alterations in individual tumors, and these can be used to design personalized evaluations for circulating tumor DNA in the serum. The same alterations have been observed in DNA from the tumor and serum samples in various types of tumors, including our result. This study suggests that the serum sample might alternatively be used when it is difficult obtaining tumor tissues for an analysis of genetic alterations. Serum sample can be obtained repeatedly and noninvasively. If the serum is used as a sample for analyzing the genetic alteration of the tumor, there will be many advantages for researchers, as well as patients.
Some studies have evaluated the relationship between the status of KRAS mutation and clinicopatholic features. Zlobec et al. [18] reported that KRAS mutation was not associated with any of the following clinicopathologic features: gender, age, tumor location, stage, and vascular invasion. However, in the study of Naguib et al. [19], KRAS mutation was closely related to advanced stage and microsatellite stability. In our study, there was no significant difference for clinicopathologic features according to the status of KRAS mutation in both the serum and tumor samples.
Whether KRAS mutation in CRC had a prognostic role independent of anti-EGFR therapies has been controversial. Our data demonstrate that KRAS tumor mutation status in both the serum and tissue samples has no major prognostic value for OS in patients with advance CRC. This finding was consistent with the data from other smaller retrospective studies [20,21]. However, simultaneously, conflicting findings were reported in the two large collaborative Kristen Ras in Colorectal Cancer Collaborative Group (RASCAL) studies of 2,721 and 4,268 patients with CRC, respectively [3,14]. Maybe, the clinical impact of our results is unstable to conclude because many other factors may affect our findings. Also, the prognosis of CRC was affected from a mutational status of many other genes, as well as KRAS. The tumorigenesis and tumor progression of CRC result from multiple genetic and epigenetic abnormalities, including defective DNA mismatch repair and mutation of KRAS, NRAS, BRAF, PI3K, PIK3CA, and p53 [22-24]. These various genetic and epigenetic changes may affect the survival of patients with CRC. However, in this study, we focused only on the status of KRAS mutation to analyze the survival of CRC.
In our study, among the 34 patients with a wild type KRAS in the tissue sample, eight revealed KRAS mutation (23.5%) in the serum sample. This discordance may be due to several reasons. First, sampling errors are responsible. Second, methods of detection for KRAS mutation are insufficient and different. Direct sequencing seems to be unable to provide satisfactory result of the status for KRAS mutation in the tissue samples containing a mixture of mutant and wild type DNA. The difference of technique applied to analyze the serum and tissue samples might affect the result for the discordance of KRAS mutation. Third, we can consider the early dissemination model of the tumor progression. In the early dissemination model, tumor cell separate from the primary lesion before the acquisition of a fully malignant phenotype to undergo new mutations and metastatic growth at the distant sites [25]. Discordance in mutation between the primary lesion and metastatic lesion may be explained by this model.
This study had some limitations, such as small number of patients, heterogeneous patients’ population, single institutional research, and the use of different techniques for analysis of both the serum and tissue. To verify the validity of the serum as a substitute for the tumor tissue, we should have used the same technique for both the serum and tissue. Also, we focused only the status of KRAS mutation in both the serum and tissue samples, although various genetic and epigenetic changes may affect the survival of patients with CRC. Nevertheless, our results suggest that the serum sample might alternatively be used when it is difficult to obtain the tumor tissue for analyzing the status of KRAS mutation status in patients with advanced CRC.
Conclusion
It is important to understand the KRAS mutation status to identify patients who might benefit from EGFR monoclonal antibody therapies and to make decisions in clinical practice. The serum sample might alternatively be used when it is difficult to obtain tumor tissues for analyzing the status of KRAS mutation in patients with advanced CRC.
 XML Download
XML Download