

 PDF
PDF Citation
Citation Print
Print


Introduction
Lung cancer is the most common cancer worldwide, comprising an estimated 12.7% of all cancer cases and 18.2% of all cancer related mortalities [1]. Lung cancer is classified histologically as either non-small cell lung cancer (NSCLC; 85%-90% of all lung cancers) or small cell lung cancer [1]. Conventional first-line therapies to treat NSCLC included a combination of premetrexed and cisplatin along with a host of other chemotherapies [2]. Although addition of bevacizumab to the first line regimen increased progression free survival, it failed to increase overall survival [3-5]. While targeted therapy with epidermal growth factor receptor (EGFR) inhibitors such as erlotinib in random patients with NSCLC resulted in modest effects [6], treatment of patients harboring EGFR mutations with erlotinib resulted in more than 60% response rates and significant progression-free survival [7,8]. The remarkable results with EGFR inhibitors in molecularly selected populations of NSCLC accelerated the quest for specific and safer targeted therapies for this dreaded disease.
Akt, also known as protein kinase B, is a serine-threonine kinase, a vital component of the phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway. It is overly expressed in most cancers [9]. Akt is activated by binding of its pleckstrin homology domain with phosphatidylinositol-3,4,5-triphosphate, produced upon phosphorylation of phosphatidylinositol-4,5-bisphosphate by PI3K [10,11]. Activated Akt phosphorylates downstream signaling molecules that include GSK3β, PRAS40, BAD, and p70s6k, resulting in survival, proliferation, growth and metastasis of cancer cells [12]. Due to its central role in these pathways, inhibition of Akt is an attractive intervention strategy for the treatment of cancer [13]. Akt exists as three isoforms with differential expression in various tissues [14]. While Akt1 is the predominantly expressed isoform controlling the growth and survival of cancerous cells, Akt2 is an important mediator of the insulin signaling pathway [14]. The role of Akt3 is still unclear; however its overexpression in brain tissue suggests its role in neuronal cells [14].
Researchers are actively targeting Akt for cancer therapy, with inhibitors such as MK-2206 and perifosine being tested in clinical trials [15]. Although, Akt isoform specific inhibitors are also reported, pan and Akt1/2 dual inhibitors are believed to be more active because of the non-redundant role of Akt1 and Akt2 in cell survival and growth [16]. Allosteric inhibitors, rather than ATP competitive inhibitors of Akt, are associated with fewer adverse effects while improving specificity for NSCLC [17]. However, poor oral bioavailability and modest clinical activity as single agents has hampered the development of several Akt inhibitors in advanced stages of clinical trials [18]. A need therefore exists for Akt specific inhibitors with acceptable drug-like properties and a higher therapeutic index for treatment of NSCLC.
A novel scaffold of Akt inhibitors were developed through virtual screening of chemical databases available at Birla Institute of Technology and Science, Pilani, Hyderabad based on docking studies using Maestro ver. 8.5 (Schrodinger, New York, NY). A benzothienopyrimidine derivative (BIA-6) was identified as a potential lead molecule based on its ability to inhibit Akt1 kinase activity with IC50 value of 256 nM [19]. In the present study, the efficacy of BIA-6 was evaluated in lung cancer cell lines. Effect of BIA-6 on downstream signaling events and its ability to synergize with the standard lung cancer therapy was also determined.
Go to : 
Materials and Methods
1. Reagents
All chemicals and reagents were purchased from Sigma (St. Louis, MO) unless otherwise indicated. Guava cell cycle reagent was purchased from Guava Technologies (Hayward, CA). Antibodies were purchased from Cell Signaling (Danvers, MA). FLICA reagent was purchased from Millipore (Billerica, MA).
2. Cell culture
Human lung carcinoma cell lines NCI-H460, NCI-H1975, NCI-H2170, and A549 were procured from American Type Culture Collection (ATCC, Manassas, VA) and grown in RPMI medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution. Human umbilical vascular endothelial cells (HUVEC) were also purchased from ATCC and cultured in F-12K medium supplemented with 10% FBS, 0.1 mg/mL heparin and 0.05 mg/mL endothelial cell growth supplement. Human tracheal epithelial cells (HTEpiC) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained in bronchial epithelial cell medium (BEpiCM). Cells were maintained at 37°C in a 5% CO2/95% air incubator and were subcultured in 1:5 ratio twice a week.
3. Cell proliferation assay
Cells were plated at 5,000 per well in a 96-well plate, treated with appropriate concentrations of BIA-6 and incubated for 72 hours at 37°C in a 5% CO2/95% air incubator. Cell viability was determined by estimating the amount of soluble formazan (in dimethyl sulfoxide) formed after addition of 100 μg MTT and a 3.5 hours incubation at 37°C. Media was removed and the crystals were dissolved in 150 μL dimethyl sulfoxide. Absorbance was measured at 560 nm on Fluostar Omega (BMG Labtech, Carry, NC).
4. Apoptosis assay
Cells were plated at 100,000 per well in a 6-well plate and incubated with the compound for 72 hours at 37°C in a 5% CO2 incubator. Cells were washed and incubated with FLICA reagent (FAM-DEVD-FMK based caspase inhibitor) for 30 minutes, and fluorescence measured at 485 nm excitation and 520 nm emission on Fluostar Omega (BMG Labtech).
5. Cell cycle analysis
Cells were plated at 100,000 per well in a 6-well plate and incubated with the compound for 72 hours at 37°C in a 5% CO2 incubator. After incubation, cells were fixed in 70% ethanol and stored at 4°C till analysis. Cells were stained with Guava cell cycle reagent (Guava Technologies) according to the manufacturer's instructions. Cell cycle data were obtained using the Guava Personal Cell Analysis system (Millipore).
6. Western blotting
Cells were plated at 200,000 per well in a 6-well plate and incubated with the compound for 4 hours. Lysates were made using RIPA buffer, run using sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane and probed with phospho-Akt (S473) antibody (Cell Signaling) followed by antirabbit IgG (Cell Signaling). Band intensity was calculated using ImageJ ver. 1.42 (National Institutes of Health, Bethesda, MD).
7. Statistical analysis
Statistical analyses were performed using GraphPad Prism ver. 6.0 (La Jolla, CA). Data were expressed as mean ± standard error of the mean (SEM). One way analysis of variance (ANOVA) was performed followed by Dunnett’s as a post hoc test. Null hypotheses of no difference were rejected if p-values were less than 0.05.
Go to :
Results
1. BIA-6 inhibits Akt dependent cellular proliferation
Expression levels of p-Akt in various cell lines were evaluated prior to using them as tools to test compounds. Rank order of p-Akt expression in the cell lines tested was H460 > A549 > H2170 > H1975 (Fig. 1). Anti-proliferative activity of BIA-6 in the aforementioned cell lines was associated with the expression levels of p-Akt (Fig. 2A). Half maximal growth inhibition (GI50) values (Table 1), indicate a ten-fold difference in sensitivity between H460 and H1975 to BIA-6. In contrast to its effect on H460 and A549, BIA-6 did not affect the proliferation of HTEpiC and HUVEC cells (Fig. 2B). A hundred-fold change in GI50 was observed between the most sensitive cell line (H460) versus normal HTEpiC cells, indicating a better selectivity of BIA-6 towards cancerous cells with inherently high levels of p-Akt.
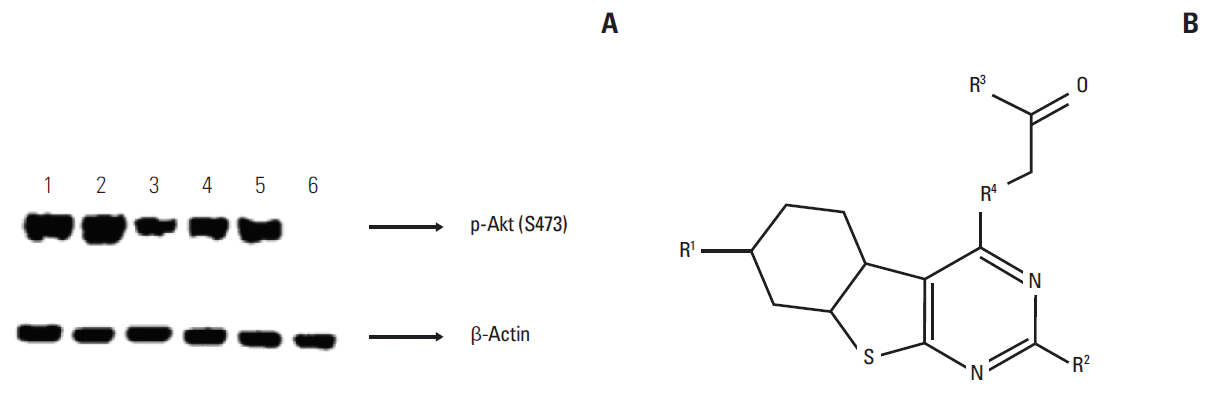 | Fig. 1.(A) Expression of p-Akt in various lung cancer cell lines. Cells were treated with BIA-6 for 4 hours, lysed and p-Akt expression was quantified by Western blot. Representation of lanes: 1, A549; 2, H460; 3, H1975; 4, H2170; 5, HTEpiC; and 6, MDA-MB-231. MDA-MB-231 was taken as a negative control cell line. (B) Markush structure for BIA-6.
|
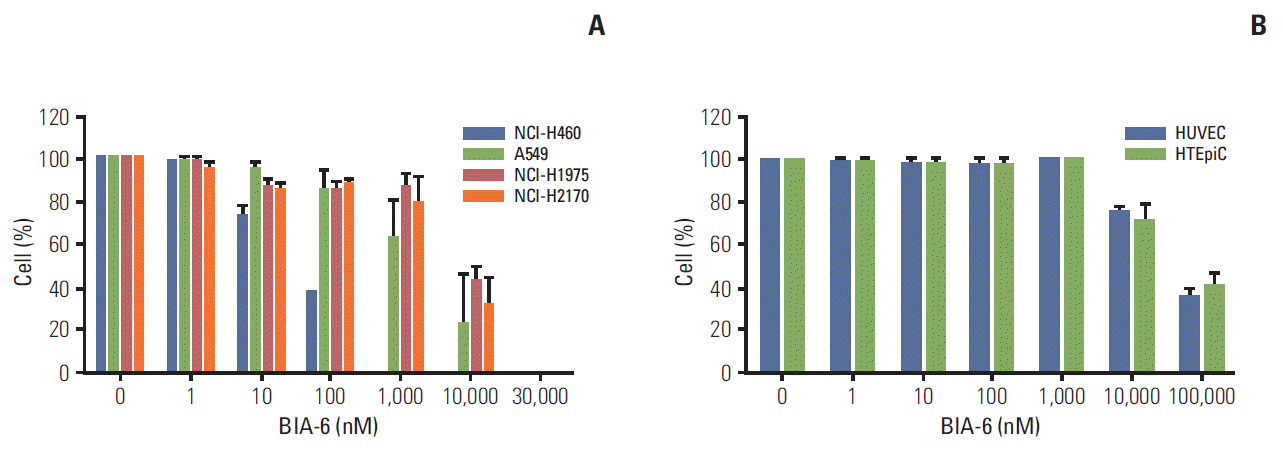 | Fig. 2.Effect of BIA-6 on cellular proliferation in lung cancer cells (A) and normal cells (B). HUVEC, human umbilical vascular endothelial cells; HTEpiC, human tracheal epithelial cell.
|
Table 1.
IC50 for BIA-6 for inhibition of proliferation and p-Akt in lung cancer cells
![]()
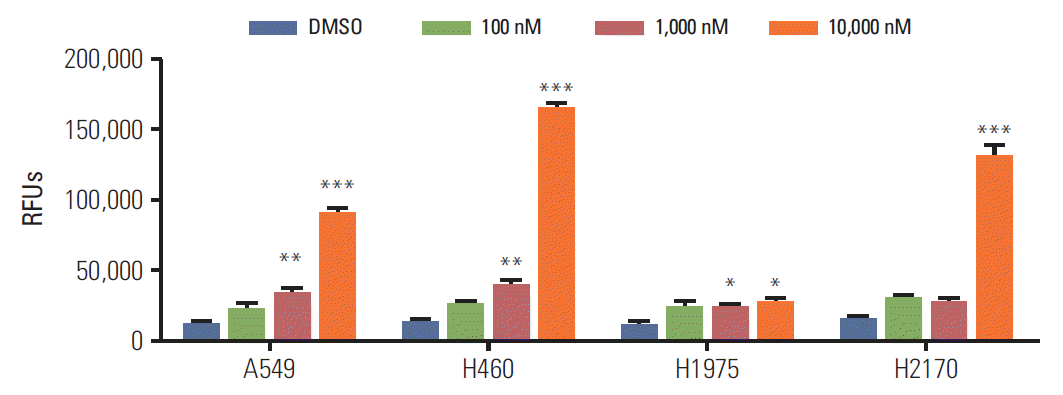
2. Caspase-3 activity in lung cancer cell lines
To evaluate the effect of BIA-6 on apoptosis, caspase-3 was measured using FLICA reagent. Fig. 3 indicates the apoptotic activity of BIA-6 at 100 nM, 1,000 nM, and 10,000 nM concentrations. There was a significant increase (p < 0.01) in the levels of caspase-3 when treated with 1,000 nM of BIA-6 in H460 and A549 indicating its sensitivity. At higher concentrations, there was a 10-fold increase in caspase-3 levels in all cell lines except H1975. Induction of caspase-3 activity by BIA-6 correlates with the amount of p-Akt present in the aforementioned cell lines, indicating the p-Akt dependent sensitivity towards BIA-6.
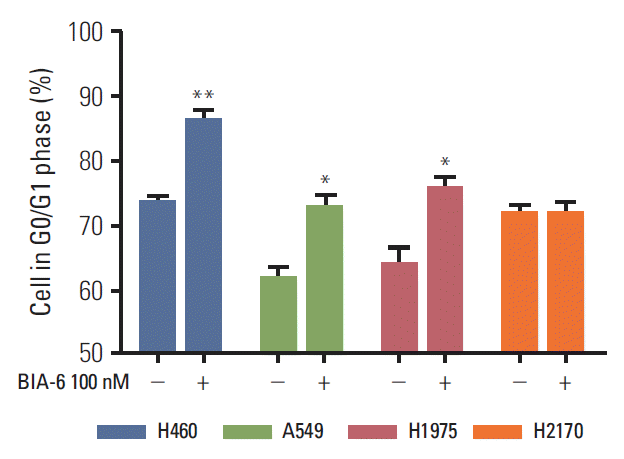
3. BIA-6 causes an arrest in the G0/G1 phase of cell cycle
The effect of BIA-6 on cell cycle in lung cancer cell lines was determined by propidium iodide staining and analyzed by flow cytometry. Fig. 4 displays the significant increase in cells in the G0/G1 phase (p < 0.01) when treated with 100 nM of BIA-6 compared to the control. At higher concentrations (1 μM and 10 μM), there was a significant increase in the % cells in sub G0 phase (p < 0.001; data not shown). Cell cycle data correlate with the levels of caspase-3 after treatment with BIA-6 in these cells. Unlike the other cell lines, G0/G1 arrest was not evident in H2170 when treated with 100 nM BIA-6.
4. Efficacy of BIA-6 in cellular assays is due to the inhibition of Akt pathway
To confirm if the activity of BIA-6 is due to inhibition of Akt phosphorylation, the lung cancer cell lines were treated with BIA-6 and the levels of p-Akt (Ser473) were determined. BIA-6 downregulated p-Akt in a dose dependent manner (Fig. 5). IC50 for BIA-6 in different cell lines are presented in Table 1. The order of sensitivity towards BIA-6 was H460 > A549 > H2170 > H1975. A forty-fold difference in activity between sensitive (H460) and resistant cell lines (H1975) was noticed upon treatment with BIA-6. Inhibition of p-Akt expression was associated with the anti-proliferative activity of BIA-6 in lung cancer cell lines, clarifying the mode of action of BIA-6.
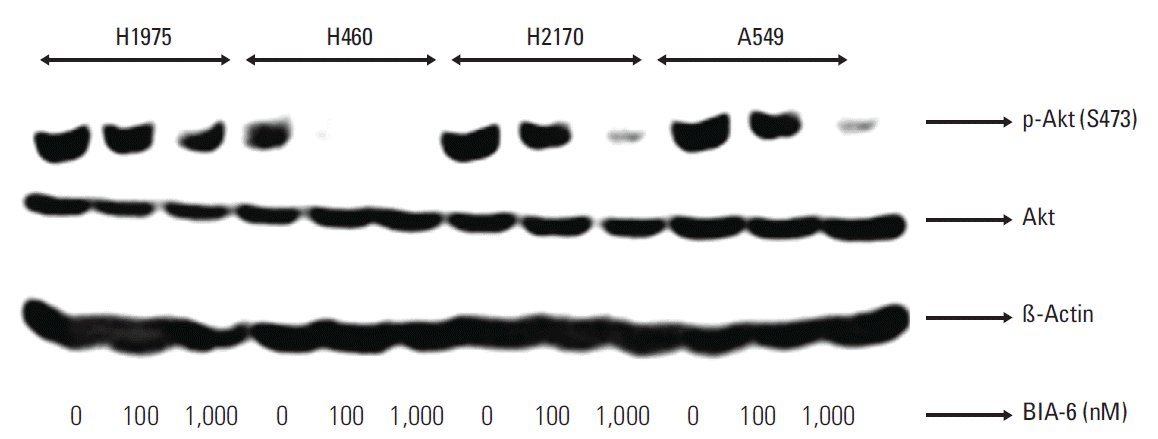 | Fig. 5.Cellular efficacy is due to the inhibition of Akt pathway. Cells were treated with BIA-6 for 4 hours, lysed and p-Akt was estimated by Western blot. The levels of p-Akt were quantified at 100 nM and 1,000 nM concentration in lung cancer cell lines. The levels of p-Akt were quantified at 100 nM concentration in lung cancer cell lines.
|
5. BIA-6 exhibits synergism with standard lung cancer therapy
Lung cancer cell lines (H460, A549, H1975, and H2170) were treated with a combination of BIA-6 and gemcitabine (GEM) or BIA-6 and docetaxel (DOC) to test for anti-proliferative activity (Fig. 6). Cells were treated at a fixed ratio of 1:10 of GEM/DOC: BIA-6 and the combination indices (CI) were calculated according to Chou and Talalay method using CompuSyn [20]. CI values were calculated on the basis of parameters derived from median effect plots of BIA-6 alone, GEM/DOC alone, and the combination of two agents at fixed ratios. A CI < 1 is evidence for synergy, whereas a CI > 1 indicates antagonism. A CI value approaching 1 indicates an additive effect. As represented in Table 2, a strong synergism was identified between BIA-6 and GEM/DOC across all the cell lines.
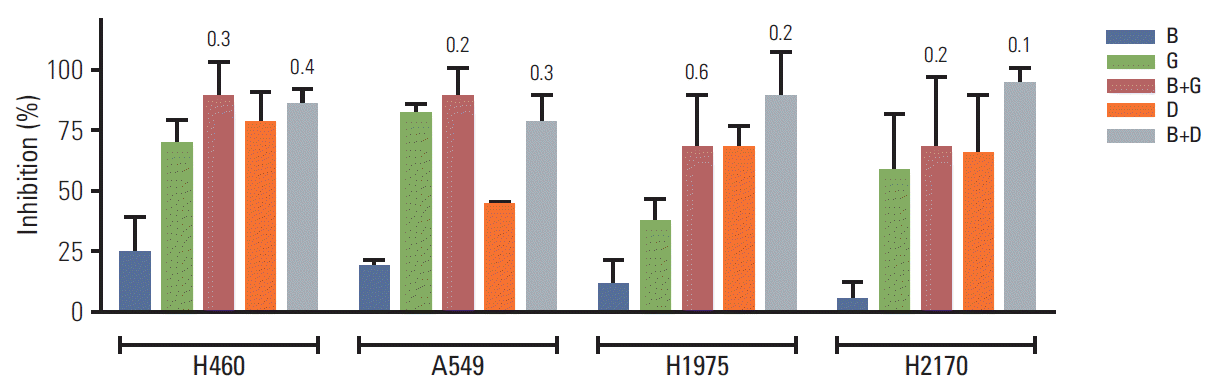 | Fig. 6.Combination of BIA-6 with gemcitabine/docetaxel in lung cancer cell lines. The concentrations of the drugs are as follows: B, BIA-6, 1,000 nM; G, gemcitabine, 100 nM; D, docetaxel, 100 nM. Values in the graph indicate the combination indices at the respective concentrations. Combination index < 1 indicates synergism.
|
Table 2.
Combination of BIA-6 (B) with gemcitabine (G)/docetaxel (D) in lung cancer cell lines
| Cell line | Combination |
Combination index (CI)a) |
|||
|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | ED95 | ||
| H460 | B+G | 0.364 | 0.280 | 0.216 | 0.182 |
| B+D | 0.132 | 0.114 | 0.103 | 0.098 | |
| A549 | B+G | 0.546 | 0.298 | 0.172 | 0.124 |
| B+D | 0.140 | 0.099 | 0.07 | 0.055 | |
| H1975 | B+G | 1.215 | 0.838 | 0.583 | 0.458 |
| B+D | 0.149 | 0.084 | 0.049 | 0.035 | |
| H2170 | B+G | 0.323 | 0.258 | 0.214 | 0.193 |
| B+D | 0.079 | 0.041 | 0.021 | 0.014 | |
![]()
Go to :
Discussion
The PI3K/Akt/mTOR signaling cascade is a pivotal pathway that is deregulated in a wide variety of human cancers and strongly contributes to both tumorigenesis and therapy resistance [21]. Considering the crucial role of aberrantly activated Akt in the pathogenesis of lung cancer, we studied the efficacy of BIA-6, a novel allosteric Akt inhibitor, as a potential therapeutic agent.
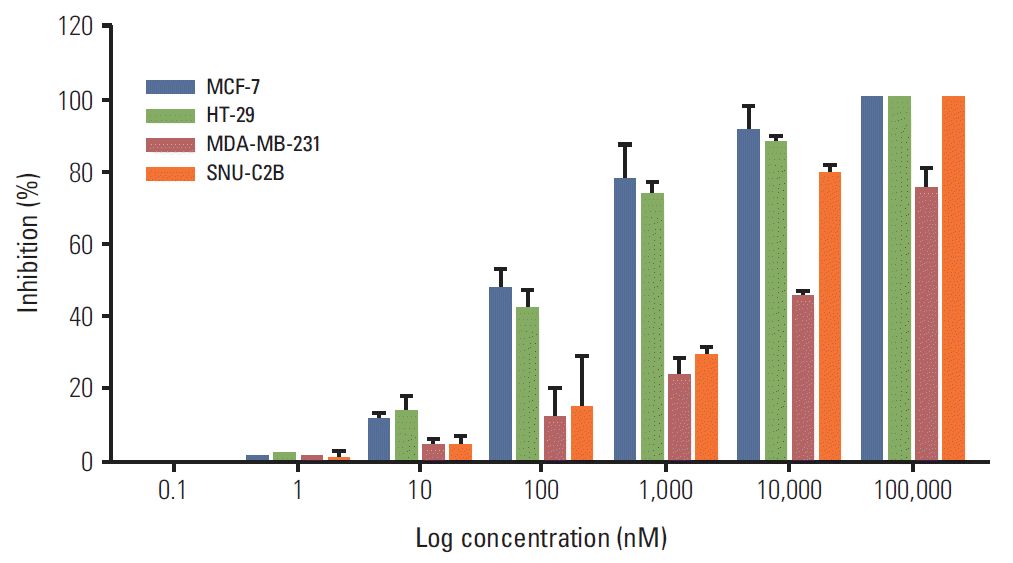
In the current study, BIA-6 is efficacious in lung cancer cell lines harboring mutations of PIK3CA (H460). The finding is consistent with the powerful role of this mutation in activation of the PI3K/Akt pathway. Significant preferentiality of the effects of BIA-6 was observed in cells harboring RAS mutations (A549). The effect of BIA-6 was minimal in cells that did not harbor PI3K/Akt mutations or RAS mutations (H1975, H2170). The genetic dependence of BIA-6 activity in lung cancer cells was reflected in lower IC50 values as well as higher levels of capsase-3 in H460 followed by A549. Similar results were observed in breast and colon cancer cell lines. Cell lines such as MCF7 and HT-29 harboring PIK3CA mutations were more sensitive to inhibition by BIA-6 compared to the ones with KRAS mutation such as MDAMB-231 and SNU-C2B (Appendices 1 and 2).
BIA-6 dephosphorylated Akt on Serine 473, indicating an indirect targeting of mTORC2, through the Akt/mTOR cascade. As Akt is upstream to TSC1/TSC2 complex, BIA-6 indirectly downregulates mTORC2 activity [22]. The efficacy of BIA-6 in inhibiting H460, A549 cell proliferation is due to its ability to both arrest the cell cycle and induce apoptosis. This was evident with the increase in caspase-3 levels in cells at lower concentrations followed by an increase in the proportion of cells in G0/G1 phase.
BIA-6 demonstrated synergism with standard chemotherapeutic agents for lung cancer (GEM/DOC) [23]. Results indicated a CI value < 1 irrespective of the genetic dependence of the BIA-6 activity. Although GEM/DOC inhibit Akt phosphorylation in cancer cell lines, they are associated with severe side-effects such as anemia, neutropenia, thrombocytopenia, infection, hair loss, nausea and diarrhea, thereby limiting their efficacy potential [24,25].
Findings from this study could have potential therapeutic implications in lung cancer patients. Combination of BIA-6 with low doses of GEM/DOC increased the cytotoxic effect of BIA-6. BIA-6 is generally well tolerated at doses required for in vitro activity and the drug only slightly affected the proliferation of normal tracheal epithelial cells (HTEpiC) and endothelial cells (HUVEC). Addition of BIA-6 to GEM/DOC could help overcome the limitation related to the therapeutic index of the chemotherapeutic agents.
Go to :
Conclusion
The current findings support the utility of BIA-6, a novel allosteric Akt inhibitor for the treatment of lung cancer. The combination of BIA-6 with standard agents clearly results in an enhanced antitumor efficacy over that seen with monotherapy. The findings provide a rationale for the clinical application of these combinations across multiple types of lung cancers.
Go to :
 XML Download
XML Download