

 PDF
PDF Citation
Citation Print
Print


Introduction
Gemcitabine, which is currently a treatment for pancreatic cancer, is used in combination with several agents, such as cisplatin, oxaliplatin, capecitabine, albumin-bound paclitaxel, or erlotinib [1-6]. Few patients with pancreatic cancer show beneficial and durable responses to gemcitabine-based chemotherapy. In an unselected population, pancreatic cancer patients who received gemcitabine-based chemotherapy had a median survival of approximately 6 months. Pancreatic cancer patients who underwent curative surgery followed by adjuvant gemcitabine chemotherapy also show poor prognosis, with a 5-year survival rate of less than 25% [6]. Therefore, proper selection of patients whose tumors are susceptible to gemcitabine therapy is important in order to maximize the benefit of chemotherapy and minimize toxicity from unnecessary chemotherapy.
The p21-activated kinase (PAK) family members, key effectors of the Rho family of GTPases, act as regulatory switches, which control cytoskeletal reorganization, gene transcription, cell proliferation and survival, and oncogenic transformation [7]. The PAK family consists of six members and is subdivided into two groups: group I (PAK1-3) and group II (PAK4-6) [8]. These kinases play a major role in oncogenic processes [7,9] and are overexpressed in many human cancers, including breast, ovarian, colorectal, thyroid, and pancreatic cancer [10]. Therefore, PAKs have been considered as potential therapeutic targets, and several inhibitors of PAKs have been developed and tested in various cancers, although no clinical data on the outcomes have been reported yet [11,12]. In addition, in a preclinical study of non-small cell lung cancer cell lines [13], a PAK1 inhibitor showed synergistic effects in combination with several anticancer agents, including oxaliplatin, erlotinib, gefitinib, lapatinib, and dasatinib.
In pancreatic cancer, it was reported that PAKs are commonly overexpressed or gene-amplified [10,14]. Oncogenic RAS, which is mutated in almost all pancreatic cancer cell lines, activates PAK1 and PAK4 [10,14]. PAK4 promotes pancreatic cancer cell motility and invasion [14]. Inversely, suppression of PAK1 by smad4 induces cell death [15]. PAK1 activation has been correlated with MUC13, a transmembrane mucin associated with pancreatic tumorigenesis [16]. Because PAKs are involved in various signaling pathways in pancreatic cancer, inhibition of these kinases could enhance drug sensitivity by altering various molecular signaling events and, when combined with chemotherapy, may represent a promising therapeutic strategy for pancreatic cancer [12]. However, the effect of gemcitabine on PAK expression in pancreatic cancer is unknown.
In this study, we attempted to determine whether the expression of major PAK isoforms could be used to predict the sensitivity of pancreatic cancer to gemcitabine and whether PAKs could be a therapeutic target in pancreatic cancer treatment.
Materials and Methods
1. Cell lines and chemicals
The Capan-1, Capan-2, MIA PaCa-2, PANC-1, Aspc-1, SNU-213, and SNU-410 pancreatic cancer cell lines were obtained from the Korean Cell Line Bank (KCLB). The cell lines have been previously described by Deer et al. [17] and Ku and Park [18]. Capan-1, Capan-2, Aspc-1, SNU-213, and SNU-410 were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). MIA PaCa-2 and PANC-1 cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS. All cells were cultured in a humidified incubator at 37°C with 5% CO2. Gemcitabine was purchased, dissolved in phosphate buffered saline, and stored at –20°C.
2. Cell viability inhibition by gemcitabine
Pancreatic cancer cell lines were seeded at 3×103 cells per well in 96-well white flat-bottomed plates. After a 24-hour incubation, cells were treated with gemcitabine at various concentrations (1 μM, 10 μM, or 100 μM) in the same type of media they were cultured in for 72 hours. Viability of these treated cells was measured by addition of 100 μL of CellTiter-Glo reagent (Promega, Madison, WI) to each well. The plates were then shaken at room temperature. Luminescence values for each well were determined using a SpectroMAX (Molecular Devices, Cincinnati, OH), and luminescence values from background wells, which contained no cells, were subtracted from values of experimental wells, which contained cells. These data were analyzed with SigmaPlot software ver. 10.0, using Logistic 3 parameter analysis to determine anticancer drug IC50 values, which is the concentration that produced 50% of the maximum cell death caused by the anticancer drug. All cell viability assays were performed in triplicate.
3. siRNA
PAK4 siRNA (siPAK4) was AccuTarget Custom Designed siRNA (Bioneer, Daejeon, Korea) and comprised the following targeting sequence: 5′-CUUCAUCAAGAUUGGCGAGTT-3′. AccuTarget Negative Control siRNA (Bioneer) was used as negative control siRNA. For transient transfections, 105 cells were transfected with 100 nM siRNA using Lipofectamine (Invitrogen, Carlsbad, CA).
4. Immunoblotting analysis
Pancreatic cancer cell lines and siRNA transfected cells were collected and lysed with cell lysis buffer (Cell Signaling, Danvers, MA). Protein concentrations of the lysate were determined using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL). Equivalent amounts of protein from each lysate were run on an sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and then transferred to nitrocellulose membranes for immunoblotting. The membranes were washed three times with Tris-buffered saline containing 0.1% Tween 20 (TBST). After blocking with 5% nonfat milk in TBST for 1 hour, the membranes were incubated with the primary antibody to PAK4 (Abcam, Cambridge, MA), human equilibrative nucleoside transporter 1 (hENT1; Proteintech, Chicago, IL), excision repair cross-complementing rodent repair deficiency, complementation group 1 (ERCC1, Santa Cruz Biotech, Santa Cruz, CA), PAK1, phosphorylated epidermal growth factor receptor, EGFR, pAkt, Akt, pErk, Erk, and β-actin (Cell Signaling). All of the primary antibodies were diluted in 3% nonfat milk in TBST and incubated at 4°C overnight. The membranes were then washed three times with TBST for 30 minutes, incubated with either a goat anti-rabbit or anti-mouse IgG HRP-linked secondary antibody diluted 1:4,000 for 2 hours at room temperature, and washed three times with TBST for 1 hour. The membranes were developed using an ECL western blotting substrate (Promega) according to the manufacturer’s instructions.
5. Reverse transcription polymerase chain reaction
Total RNA was extracted from cells using Trizol reagent (Invitrogen) and reverse-transcription into single-stranded cDNA was performed using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s recommendations. The primer sequences and PCR conditions for detection were as follows: β-actin-F 5'-AGTGTGACGTTGACATCCGT-3', β-actin-R 5'-GCAGCTCAGTAACAGTCCGC-3' (235 bp fragment), hENT1-F 5'-TCAGCCCACCAATGAAA-3', and hENT1-R 5'-GGCCCAACCAGTCAAAGATA-3' (215 bp fragment). The amplification conditions consisted of an initial denaturation at 95°C for 5 minutes, followed by 40 cycles of denaturation at 95°C for 30 seconds, annealing at 58°C for 30 seconds, and elongation at 72°C for 30 seconds. A 1% agarose gel containing Loading Star (DyneBio Inc., Seongnam, Korea) for visualization was run in a Tris-Borate-EDTA buffer for 20 minutes at 100 V, and PCR products were analyzed using a Bio Image Analyzer (Fisher Scientific).
Results
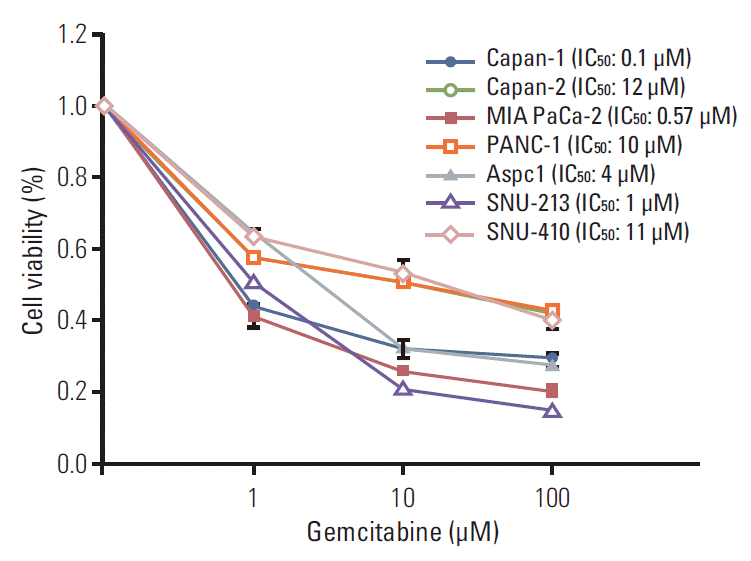
1. Sensitivity of human pancreatic cancer cell lines to gemcitabine as measured by cell viability
As shown in Fig. 1, the cytotoxic effect of gemcitabine on the seven human pancreatic cancer cell lines (Capan-1, Capan-2, MIA PaCa-2, PANC-1, Aspc-1, SNU-213, and SNU-410) was examined using a luminescence assay. The IC50 values for gemcitabine were 0.1 μM, 12 μM, 0.57 μM, 10 μM, 4 μM, 1 μM, and 11 μM in Capan-1, Capan-2, MIA PaCa-2, PANC-1, Aspc-1, SNU-213, and SNU-410 cells, respectively. The cell viability of the Capan-2, PANC-1, and SNU-410 cell lines reflected increased resistance to gemcitabine, which was defined as IC50 ≥ 10 μM gemcitabine. The other cancer cell lines (Capan-1, MIA PaCa-2, Aspc-1, and SNU-213) showed relatively good sensitivity to gemcitabine.
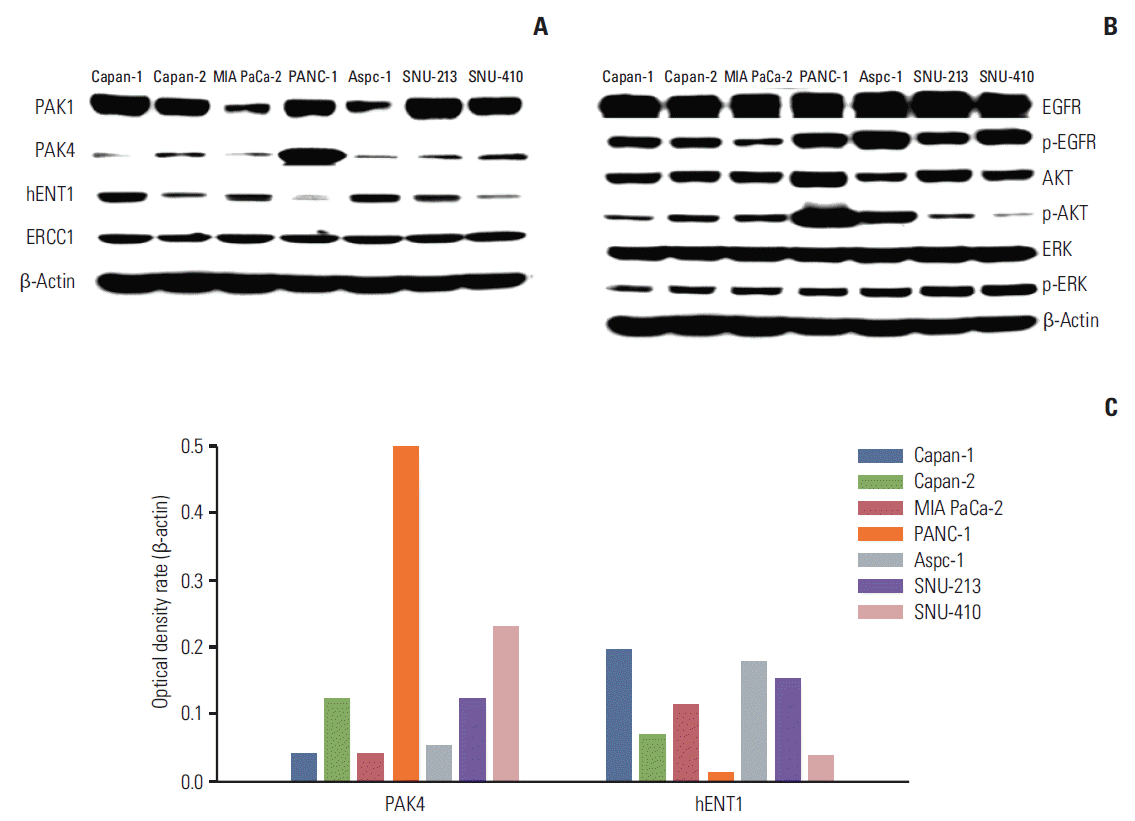
2. Protein expression of PAK isoforms and signaling molecules related to gemcitabine sensitivity
The protein expression levels of PAK1, PAK4, hENT1, or ERCC1 in the seven human pancreatic cancer cell lines were assessed by immunoblotting (Fig. 2A). Although all of the cancer cell lines had detectable levels of PAK1, PAK4, ERCC1, and hENT1, higher levels of PAK4 protein and lower levels of hENT1 protein were seen in Capan-2, PANC-1, and SNU-410 cells, which were the cell lines that showed increased resistance to gemcitabine (Fig. 1). Inverse correlation was observed between PAK4 expression and hENT1 expression. Expression of PAK1 and ERCC1 did not show correlation with cell sensitivity to gemcitabine. In addition, no correlation was observed between expression of PAK isoforms and expression of ERCC1 protein was evident. In the EGFR/ERK/AKT signaling pathway (Fig. 2B), no signaling molecules showed correlation with gemcitabine sensitivity. The effects of PAK isoforms on EGFR, ERK, and AKT were not consistent. Correlation between PAK4 expression and hENT1 expression was determined using densitometry (Fig. 2C).
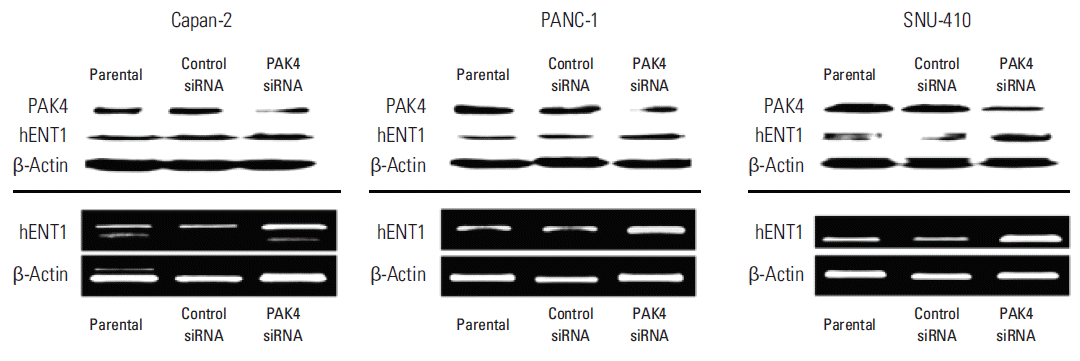
3. Expression of hENT1 following downregulation of PAK4
We found an inverse correlation between PAK4 and hENT1 protein expression levels. To evaluate the relationship further, PAK4 expression was knocked down using siRNA in the three gemcitabine-resistant pancreatic cancer cell lines, which showed higher PAK4 expression. Expression of hENT1 in PAK4 knockdown pancreatic cancer cell lines was detected by immunoblot and reverse transcription polymerase chain reaction (RT-PCR) (Fig. 3). PAK4 protein levels were reduced in cells treated with PAK4 siRNA compared to parental cell lines or lines treated with control siRNA (Fig. 3, upper panel). Inversely, PAK4 siRNA induced slightly higher expression of hENT compared with that in parental cell lines or lines treated with control siRNA. To confirm the higher expression of hENT1 after PAK4 knockdown, RT-PCR analysis of hENT1 was performed (Fig. 3, lower panel). In these assays, a higher level of hENT1 mRNA was observed in cells treated with PAK4 siRNA compared to that in parental cell lines or lines treated with control siRNA. Therefore, our results indicate that downregulation of PAK4 would induce upregulation of hENT1 in gemcitabine-resistant pancreatic cancer cells.
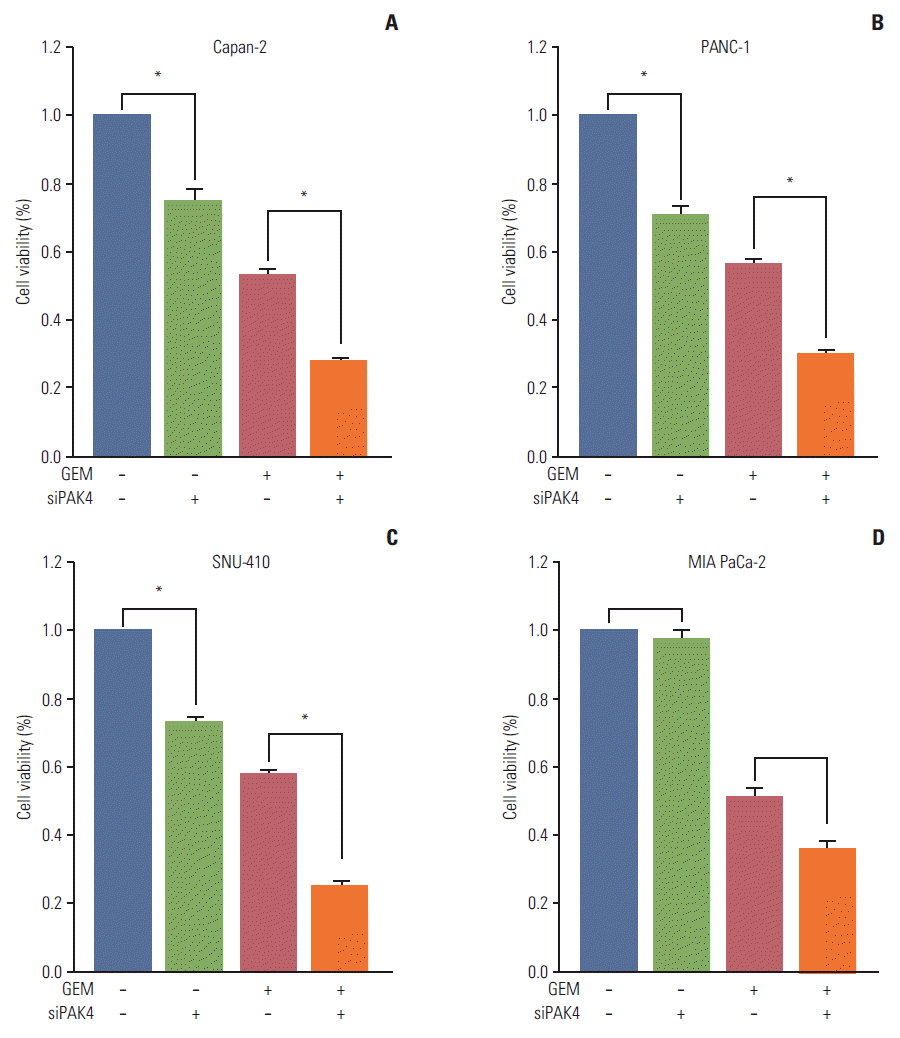
4. Cell sensitivity to gemcitabine following PAK4 knockdown
To determine the potential of PAK4 as a therapeutic target, cell viability assays of PAK4 knockdown cells were performed in gemcitabine-resistant pancreatic cancer cell lines along with one cell line with lower PAK4 expression, MIA PaCa-2. PAK4 knockdown resulted in an approximate 27% reduction of cell viability in cell lines with higher PAK4 expression. However, PAK4 knockdown had little effect on the MIA PaCa-2 cell line (Fig. 4). In addition, PAK4 knockdown increased the sensitivity of gemcitabine-resistant cells to treatment with the IC50 concentration of gemcitabine, inducing reduced viability over gemcitabine treatment alone. Addition of PAK4 knockdown to gemcitabine induced an approximate 50% reduction of cell viability compared with gemcitabine alone, which could be synergistic considering an approximate 27% reduction of cell viability in PAK4 knockdown alone. In MIA PaCa-2 cells, which have lower PAK4 expression, PAK4 knockdown in combination with the IC50 concentration also decreased cell viability when compared with gemcitabine alone. Therefore, knockdown of PAK4 expression could increase the sensitivity to gemcitabine by upregulating hENT1 expression.
Discussion
To the best of our knowledge, this is the first study to suggest the association between PAK4 expression and sensitivity to gemcitabine in pancreatic cancer. In our study, pancreatic cancer cell lines with a higher expression of PAK4 showed increased resistance to gemcitabine compared to those with a lower expression of PAK4, suggesting that this protein could be a predictive marker for gemcitabine sensitivity. Higher expression of PAK4 was associated with lower expression of hENT1, which is required for gemcitabine to cross the cancer cell membrane. PAK4 knockdown via PAK4 siRNA induced the upregulation of hENT1, as determined by immunoblotting and RT-PCR, suggesting that PAK4 could be a therapeutic target for control of hENT1 expression for increasing sensitivity to gemcitabine. In addition, knockdown of PAK4 in gemcitabine-resistant pancreatic cancer cell lines, which have higher PAK4 expression, resulted in a reduction in cell viability. Combination of PAK4 knockdown with gemcitabine resulted in restoration of sensitivity to gemcitabine in the gemcitabine-resistant pancreatic cancer cell lines.
PAK4 is overexpressed in various cancer cells, and many reports have previously shown that this overexpression promotes cell survival as well as anchorage-independent growth, which are important hallmarks of oncogenic transformation [19]. Increased expression of PAKs protects cells from different apoptotic stimuli, including radiation, serum deficiency, and tumor necrosis factor-α–induced cleavage of poly(ADP-ribose) polymerase and caspase-3 [20,21]. In gastric cancer, overexpression of PAK4 appears to correlate with a poor prognosis [22]. In our study, pancreatic cancer cell lines that overexpress PAK4 also showed increased resistance to gemcitabine. In our study, although expression of PAK1 was detected in pancreatic cancer cell lines, there was no association with sensitivity to gemcitabine.
Some biomarkers for prediction of gemcitabine efficacy have been identified in retrospective studies [23,24]. However, no biomarkers have been validated prospectively. Of the suggested biomarkers, hENT1, a nucleoside transporter protein required for gemcitabine to cross the cell membrane, may be a reliable biomarker [24]. hENT1 was recently reported to be a predictive marker of benefit from gemcitabine therapy in patients with pancreatic cancer who underwent surgery followed by gemcitabine chemotherapy [24]. In our study, in concordance with previous reports, pancreatic cancer cell lines with higher expression of hENT1 showed a higher sensitivity to gemcitabine. Expression levels of PAK4 and hENT1 in pancreatic cancer cell lines showed inverse correlation. Knockdown of PAK4 induced upregulation of hENT1, which resulted in increased sensitivity to gemcitabine. Therefore, the combination of PAK4 knockdown and gemcitabine treatment could be an effective strategy in treatment of pancreatic cancer with gemcitabine. In our study, the effect of gemcitabine on pancreatic cancer cell lines knocked down for PAK4 was very potent.
Several inhibitors of PAKs have been developed, and preclinical studies with PAK inhibitors have been reported [11]. Among the various inhibitors, a pyrroloaminopyrazole-based PAK inhibitor developed by Pfizer (PF-3758309) was a promising novel agent. PF-3758309 suppressed the growth of several human cancers in mice. An early phase clinical trial using PF-3758309 in patients with advanced solid tumors opened in 2009. However, this clinical trial was terminated prematurely in 2011 because of the undesirable pharmacokinetic characteristics and the lack of an observed dose-response relationship. A PAK4 inhibitor could not be used to confirm the result seen with knockdown by siRNA in our study. Although the use of PAK inhibitors was planned in our study, selection and acquisition of PAK inhibitors was difficult. This point is a limitation in our study.
Conclusion
Pancreatic cancer cell lines with higher PAK4 expression showed increased resistance to gemcitabine. Expression of PAK4 showed inverse correlation with that of hENT1. PAK4 knockdown induced upregulation of hENT1 and increased sensitivity to gemcitabine in pancreatic cancer cell lines. Therefore, the expression of PAK4 could be a predictive marker of gemcitabine sensitivity and a potential therapeutic target for pancreatic cancer. Conduct of further study for development of a PAK4 inhibitor is warranted.
 XML Download
XML Download