

 PDF
PDF Citation
Citation Print
Print


Introduction
The most common testicular neoplasms are germ cell tumors, either seminomas or nonseminomatous germ cell tumors. Spermatocytic seminoma (SS) is a rarer germ cell tumor that accounts for approximately 1%-2% of all testicular tumors and 3%-7% of seminomas. SS is an indolent neoplasm, characterized by long duration of symptoms, presentation at early stages, and low risk of distant metastasis [1]. Metastatic SS constitutes less than 0.5% of metastatic germ cell tumors and has a biological course that is rarely complicated by sarcomatous transformation [2], which is clinically characterized by a rapid increase in tumor size and higher possibility of metastatic disease [3]. The sarcomatous component of SS is presented mostly as rhabdomyosarcoma, as well as some others [4]; all of which are known to be resistant to platinum-based chemotherapy.
Metastatic SS with sarcomatous transformation is extremely rare, with just seven cases clearly documented in the literature to date, and is reported to be highly resistant to cytotoxic chemotherapy with only a 5-month median survival after diagnosis [5]. We here present a first case report of SS with extensive rhabdomyosarcomatous transformation and multiple metastatic lymphadenopathies that showed a promising response to platinum-based chemotherapy.
Case Report
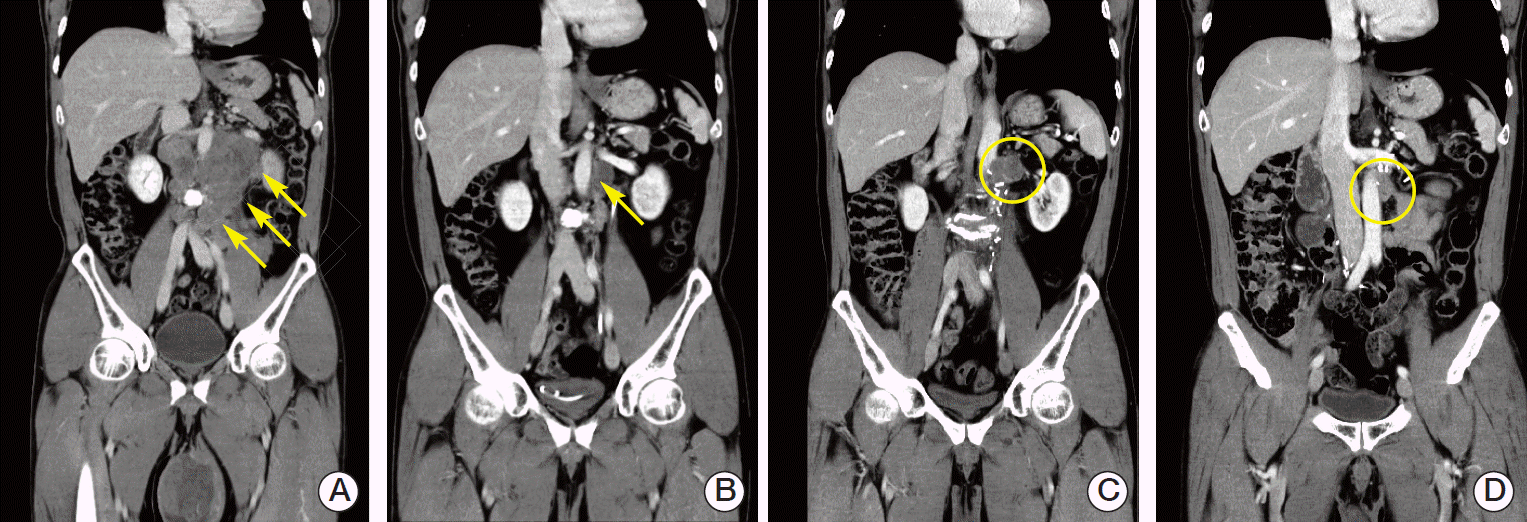
A 52-year-old male was presented at our hospital in March 2012 with complaints of severe pelvic pain and a palpable neck node, which had started 3 months previously. The patient had a large left testicular mass that had been growing slowly for 3 years before this presentation. Before undergoing an orchiectomy, this testicular mass was 9 cm in size. Tumor markers (alpha-fetoprotein and betahuman chorionic gonadotropin) were within normal limits, but his lactate dehydrogenase was elevated to 514 IU/L (normal range, 120 to 250 IU/L). Computed tomography imaging revealed multiple conglomerated lymph nodes in the left neck level V, retroperitoneal, and left common/external iliac spaces with an invasion of the left ureter by the retroperitoneal lymph nodes (the largest measuring 11 cm at the left para-aortic area), causing hydronephrosis.
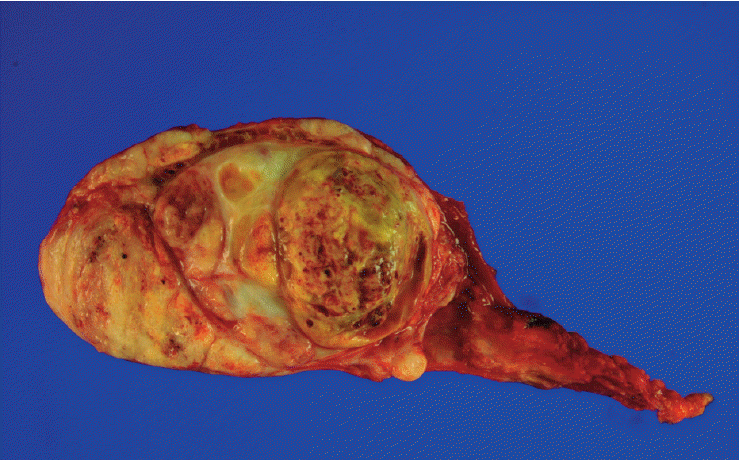
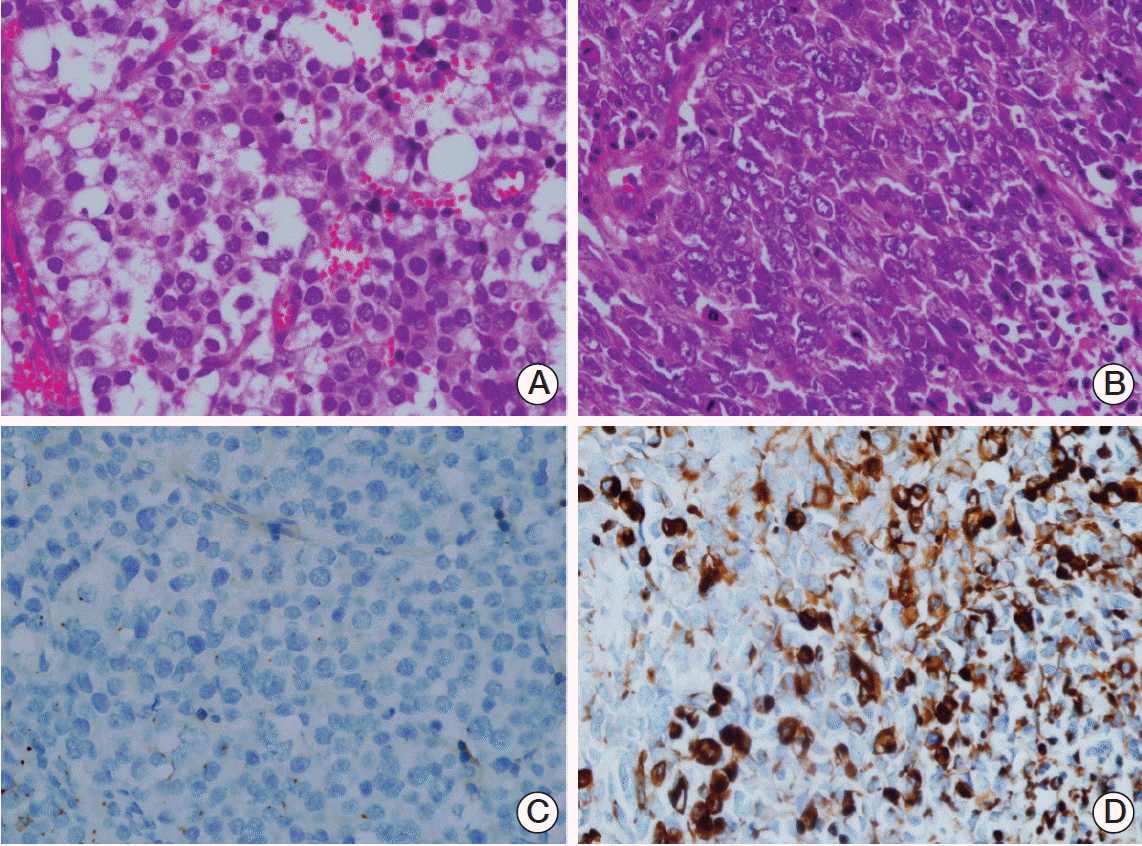
The patient underwent a left radical orchiectomy. The orchiectomy specimen was a grossly well-defined lobulated soft mass, which extended to the tunica albicans and epididymis and was histologically revealed as SS with massive rhabdomyosarcomatous transformation (Fig. 1). An ultrasound-guided neck biopsy was performed and the histological features of the lymph node were similar to those of the testicular mass, which suggests metastatic rhabdomyosarcoma. A hematoxylin and eosin stain revealed cellular features of SS (Fig. 2A) and high-grade alveolar rhabdomyosarcoma with focal dense pink cytoplasm (Fig. 2B). Desmin and myogenin staining were absent in the SS component (Fig. 2C), but were strong in the rhabdomyosarcoma component (Fig. 2D), which occupied 70% of the tumor mass.
Immediately after the orchiectomy, the patient received first-line chemotherapy with a combination of etoposide, ifosfamide, and cisplatin (VIP). After four cycles of VIP, computed tomography imaging revealed a marked decrease in the size of the left para-aortic lymph node (3 cm) (Fig. 3B) and ablation of pelvic or neck nodes. Retroperitoneal lymph node dissection (RPLND) was recommended due to disease activity on positron emission tomography imaging. However, the patient was initially reluctant to undergo surgical resection. Unfortunately, but as expected, retroperitoneal lymph node (aortocaval and left para-aortic lymph nodes) involvement was found to have progressed on a follow-up computed tomography, and the patient finally consented to surgery. RPLND was attempted but complete resection was not possible due to adherence of the lymph nodes to the renal vessel and aorta (Fig. 3C). The pathology revealed SS with rhabdomycosarcomatous transformation.
Postoperatively, the residual lymph node involvement progressed, and second-line chemotherapy with a combination of paclitaxel, ifosfamide, and cisplatin (TIP) was administered. The patient achieved a near complete response after four cycles of TIP (1.5-cm-sized remnant left para-aortic lymph node) (Fig. 3D). For curative purposes, we considered re-operation or autologous stem cell transplantation for the residual tumor, but the plan was frustrated by its unresectability and the failure of peripheral blood stem cell collection. As an alternative, the patient underwent radiotherapy to the remnant left para-aortic lymph nodes (total, 60 Gy). At the time of writing, he remains under careful observation with no evidence of disease.
Discussion
First described by Masson in 1946 [6], SS is a rare testicular germ cell tumor with well-defined histopathological features. It is characterized by an indolent course with a long duration of symptoms, such as slow painless testicular enlargement for several years, extremely low metastatic potential, and a good prognosis. However, sarcomatous transformation, which occurs in approximately 6% of cases, triggers a sudden rapid increase in the size of the tumor and may result in poor prognosis [3,7].
In our present case, the patient had a gradual testicular enlargement for 3 years, followed by a rapidly expanding neck node for 3 months. Histological analysis revealed a high-grade rhabdomyosarcoma. Hypotheses have been previously proposed to explain the histogenesis of sarcomatous elements and their relationship to SS. One of the most plausible theories is that sarcomatous transformation is an “anaplastic transformation” or “dedifferentiation” of a well-differentiated SS. The characteristic course of this tumor is slow enlargement of the testis initially, followed by a rapid increase in size, and it favors sarcomatous dedifferentiation as the underlying mechanism [8].
The treatment of choice for non-metastatic SS is orchiectomy, followed by surveillance. Adjuvant chemotherapy or radiotherapy is rarely recommended in SS cases due to low risks of relapse and metastasis. However, a sarcomatous component in a non-metastatic disease confers a much higher risk of metastasis, and precise treatment modalities have yet to be established in these cases. Only four cases of localized SS with sarcomatous transformation have been reported thus far and none of the affected patients showed recurrence of distant metastasis after radical orchiectomy [5]. This suggests that this treatment approach was appropriate even in the presence of a sarcomatous component.
However, if metastases develop in SS cases, the prognosis is very poor, even with the aggressive multimodal approach [2]. In the iterature, five such patients had received platinum-based chemotherapy, and all responded poorly and died shortly (within 3 to 14 months) after diagnosis (Table 1) [9-11]. However, we report a first case of SS with extensive rhabdomyosarcoma transformation and with huge cervical and retroperitoneal lymphadenopathy, which responded dramatically well to the initial VIP combination chemotherapy after radical orchiectomy. The cervical lymph node was ablated and the retroperitoneal lymph node shrank to a residual size of 3 cm.
Post-chemotherapy surgery was attempted in our current patient, which revealed only a rhabdomyosarcoma component; however, complete resection could not be achieved due to its severe adhesion to the adjacent organs, including the major vessels. Shortly after RRLND, there was a progression of the residual disease. However, salvage chemotherapy with TIP regimen produced a dramatic response, with a residual tumor reduction to 1.5 cm. After radiotherapy for the residual retroperitoneal lymph node, the patient achieved complete response and remains free from disease progression at the time of writing this report. Therefore, we feel that although there is no sound rationale for consolidative radiotherapy to treat residual tumors in seminoma [12], based on the pattern of disease progression, such as increase in size and invasion of adjacent organs at the existing sites rather than new metastases, adequate loco-regional disease control by radiotherapy may be beneficial in treating SS with rhabdomyosarcoma differentiation when surgical resection fails.
Based on the outcomes in our current case with metastatic SS with rhabdomyosarcomatous transformation, a cisplatin-based chemotherapeutic approach with surgical resection of the residual disease can be effective. The mechanism underlying this unusual and dramatic response to platinum-based chemotherapy is not clear. Furthermore, cooperative research on adequate therapies and prognoses is therefore needed for this extremely rare disease.
 XML Download
XML Download