

 PDF
PDF Citation
Citation Print
Print


Introduction
Pulmonary mucosa-associated lymphoid tissue (MALT) lymphoma is a rare disease accounting for less than 1% of all non-Hodgkin lymphoma [1]. Although pulmonary MALT lymphoma has an indolent nature with a good prognosis, most patients are treated with surgery, chemotherapy, or radiotherapy [2]. The most frequent radiologic finding of the disease is bilateral consolidations with bronchus sign. Sometimes, the diagnosis of pulmonary MALT lymphoma is difficult because of atypical radiologic findings and unusual clinical manifestations [3]. Here, we report a case of pulmonary MALT lymphoma presenting with ground-glass opacities with septal thickenings in both lungs, which resolved rapidly without any treatment for the underlying malignancy.
Case Report
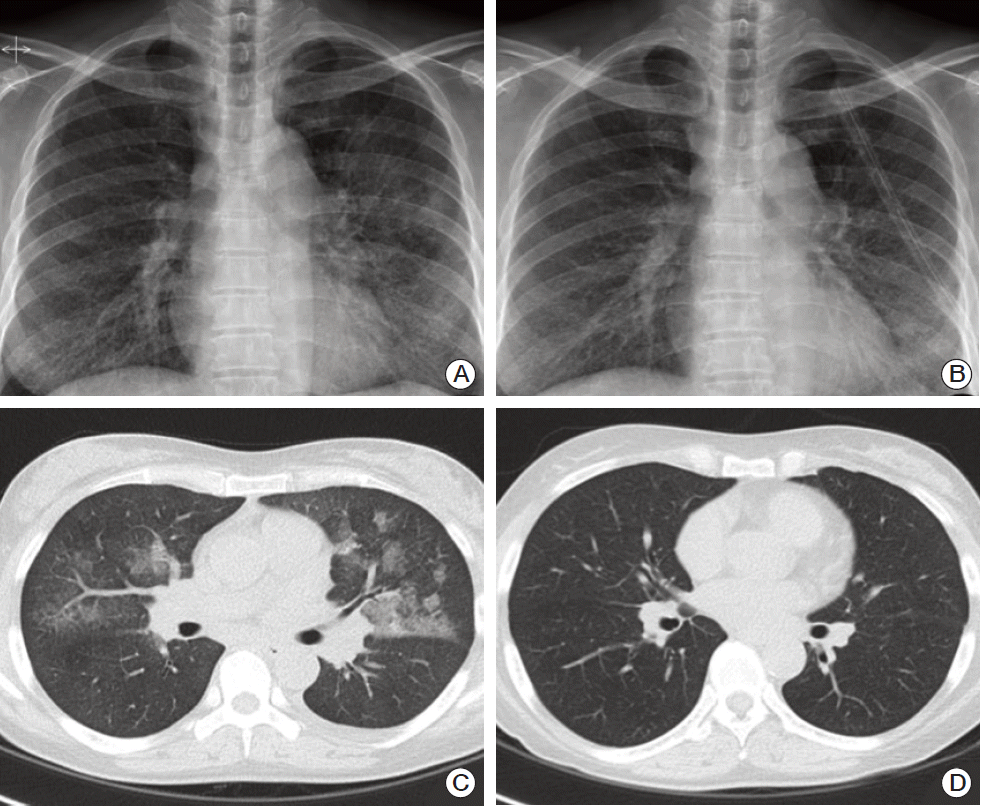
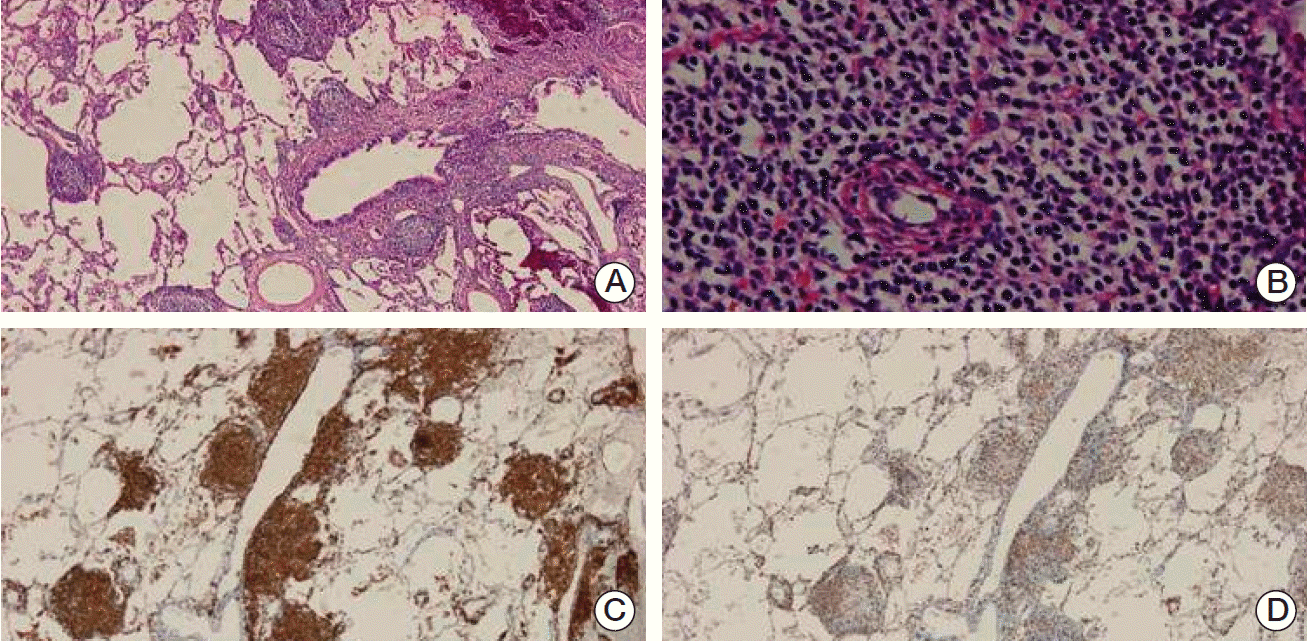
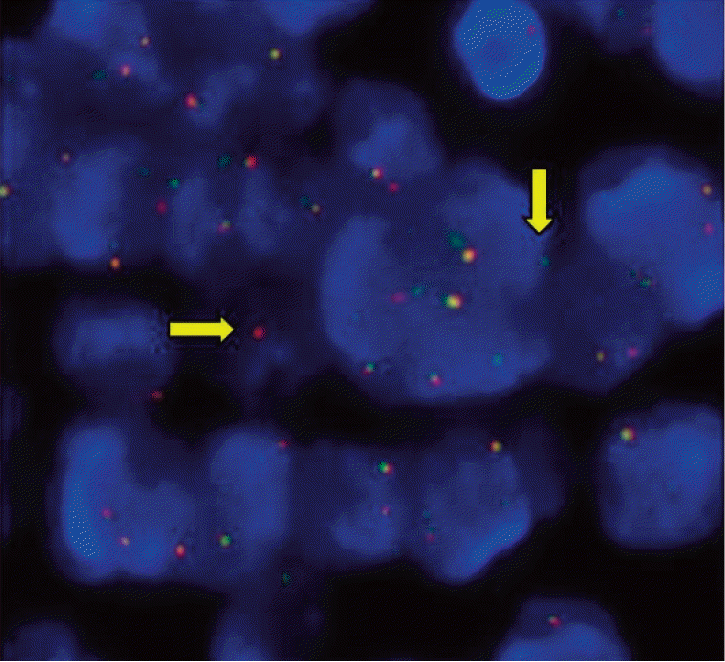
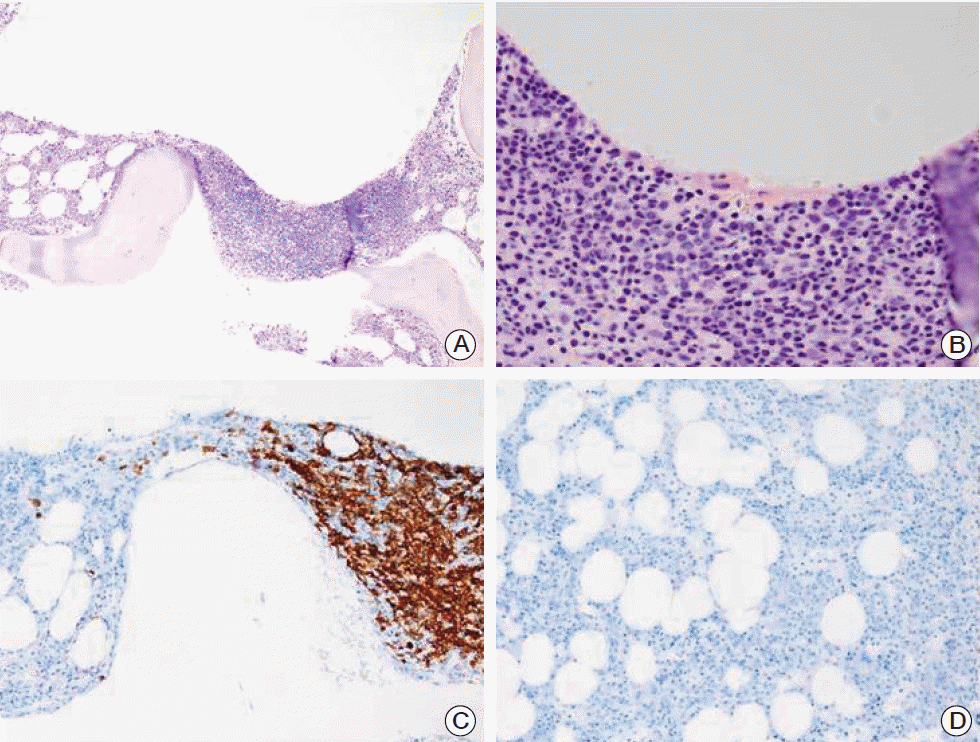
A 57-year-old woman presented to our hospital with complaints of cough, febrile sensation and dyspnea for two months. Chest X-ray and computed tomography (CT) of the chest showed multifocal ground-glass opacities in both lower lobes with interlobular septal thickenings (Fig. 1A and C). Laboratory data were within normal limits except mild elevation of C-reactive protein and serum lactate dehydrogenase, which were 5.42 mg/dL and 498 U/L, respectively. Autoantibodies for connective tissue disease-associated interstitial lung disease were all negative. Arterial blood gas analysis showed pH 7.478, pco2 32.6 mm Hg, po2 60.1 mm Hg, and SaO2 91.1% while breathing ambient air. We empirically gave her antibiotics, ceftazidime and levofloxacin. During bronchoscopy, there was no endobronchial lesion, and bronchoalveolar lavage fluid analysis showed a composition of 82% lymphocytes, 13% neutrophils, and 2% eosinophils. Microbiologic study including respiratory virus, bacteria, Mycobacterium tuberculosis, and fungus showed Acinetobacter baumannii. Follow-up chest CT after the antibiotics treatment 10 days later showed little interval change of the lung lesion. Therefore, diagnostic open lung biopsy via thoracoscopy was done at the lingular segment of the left upper lobe. Histopathology demonstrated atypical lymphoproliferative lesion on hematoxylin and eosin stain. The atypical lymphocytes were positive for CD20 and CD3 immunohistochemically (Fig. 2) and polymerase chain reaction assay for IgH gene showed monoclonality. Initially, lymphoid follicular hyperplasia was diagnosed pathologically. Her symptoms and radiologic findings (Fig. 1B) improved after open lung biopsy, so we decided to observe her at the outpatient clinic. Two months later, fluorescent in situ hybridization analysis detected API2-MALT1 translocation at the level of 80.9%, but not for MALT1 rearrangement (Fig. 3) and pulmonary MALT lymphoma was finally confirmed. At that time, she did not complain of any symptoms and follow-up chest CT showed complete regression of the previous lung lesion without any treatment (Fig. 1D). However, in a staging workup including abdomen CT, positron emission tomography-CT, esophagoduodenoscopy, and bone marrow biopsy, there was involvement of MALT lymphoma in the bone marrow. Therefore, she received chemotherapy including rituximab, cyclophosphamide, doxorubin, and vincristine. At 3 months of treatment, follow-up bone marrow aspirate demonstrated no evidence of lymphoma (Fig. 4).
Discussion
Pulmonary MALT lymphoma is a rare disease accounting for less than 1% of all non-Hodgkin lymphoma. The stomach is the most common site of MALT lymphoma. Non-gastric MALT lymphoma is not common and occurs in the intestinal tract, skin, and lung in order of frequency [1,2]. The pathogenesis of pulmonary MALT lymphoma is not known clearly, but it is supposed that long-term exposure to various antigenic stimuli, such as smoking, chronic mycobacterial infection, and autoimmune diseases including Sjögren syndrome and systemic lupus erythematosus, causes pulmonary MALT lymphoma [4]. Chronic antigen stimulation-induced inflammation and T cells seem to cooperate in triggering tumorigenesis [5].
The diagnosis of pulmonary MALT lymphoma is difficult in cases displaying non-specific clinical manifestations and atypical radiologic findings [6]. CT findings of pulmonary MALT lymphoma present diverse patterns and commonly reveal multiple, bilateral consolidations with bronchus sign. In our case, chest CT findings showed a pattern of diffuse interstitial lung disease. Diffuse interstitial lung disease pattern presenting ground-glass attenuation is not a frequent finding with an incidence of 6%-10% in pulmonary MALT lymphoma [6,7]. The imaging-histology correlation studies in pulmonary MALT lymphoma showed that whereas mass, nodule, and consolidation in the CT were caused by alveolar space filling, ground-glass opacity was caused by lymphomatous infiltration of interlobular septa and alveolar walls [6].
Pathologic diagnosis presents problems for the pathologist due to the heterogeneous appearance and similarity to reactive lymphoid tissue in the lung [8]. In such cases, polymerase chain reaction assay for IgH gene rearrangement is used to detect the monoclonality because there is evidence of monoclonality in lymphoid infiltrates [9]. However, this method has only approximately 60% sensitivity. Therefore detection of API2-MALT1 fusion in the cytologic specimen is widely used to increase diagnostic sensitivity because this gene alteration is specific to MALT lymphoma [10].
Pulmonary MALT lymphoma has an indolent nature with the potential for spontaneous regression [2]. Troch et al. [11] retrospectively investigated 11 patients undergoing a watch-and-wait policy. For the follow-up of median 28 months, six of 11 patients experienced spontaneous regression but not complete remission of the MALT lymphoma in the lung [11]. There is only one case report that documented spontaneous complete remission of pulmonary MALT lymphoma in Japan [12]. In that case, spontaneous regression was found 16 days after the tumor biopsy and relapse of the lung lesion was not detected until 20 months after the first visit as in our case. The mechanism of spontaneous regression of MALT lymphoma is not known, but in some reports, the anti-inflammatory effect of macrolide-induced apoptosis of lymphocytes via downregulation of overexpressed Bcl-xL, anti-apoptotic gene, is suggested [13,14].
In the present case, atypical chest CT findings such as diffuse interstitial lung disease pattern, initial obscure pathology, and the clinical course of rapid spontaneous regression of the lesion together made the diagnosis more difficult. Spontaneous regression of the lung lesion is not explained completely. one possible hypothesis is that the chronic respiratory inflammation was controlled by antibiotics, even though the antibiotics used were not macrolide and the timing of the radiologic resolution did not correlate with the effect of the antibiotics. After resolution of the primary lesion, selection of treatment modality was difficult. Although pulmonary MALT lymphoma has an indolent clinical course, the relapse rate is constant at approximately 35% [1]. In addition, dissemination to bone marrow is associated with poor outcomes [15]. Therefore, the patient received chemotherapy and the lymphoma infiltration in bone marrow resolved after 3 months of chemotherapy.
In conclusion, pulmonary MALT lymphoma is a rare disease and can be misdiagnosed because of atypical clinical manifestations and radiologic findings. Definite diagnosis of pulmonary MALT lymphoma requires a detailed histologic examination of the lung lesion. observation could be one of the options because the lung lesion can resolve spontaneously like our case. However, close monitoring is needed and treatment should be considered in the case of disseminated disease.
 XML Download
XML Download