

 PDF
PDF Citation
Citation Print
Print


Introduction
Many strategies shown to be effective in the chemoprevention of cancer or the precancerous stage change the signaling pathways targeted by therapies currently being used in the lung; however, no drug effective as a lung cancer chemotherapy agent has been identified to date [1]. Accordingly, prevention of lung cancer may be a better approach, and the use of multiple agents that target different molecules involved in carcinogenesis holds great promise [2,3]. Several studies have shown that low-dose administration of a combination of cancer preventive agents with different modes of action may produce synergistic effects on efficacy and minimize possible side effects associated with high-dose administration.
Non-steroidal anti-inflammatory drugs (NSAIDs) have potential chemotherapeutic efficacy against carcinogenesis that occurs via inhibition of cyclooxygenase (COX) enzyme [4], which catalyzes the production of prostaglandins from arachidonic acid and therefore plays an important role in inflammation and cancer [5]. There is accumulating evidence of the cancer preventive effects of NSAIDs. However, the relatively high doses required for the observed chemopreventive effect in human trials may discourage the long-term use of NSAIDs for lung cancer prevention owing to the possibility of increased risk of serious gastrointestinal and cardiovascular side effects [6,7]. An alternative approach is to combine NSAIDs with other chemopreventive agents.
Statins, or 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are a class of drugs that inhibit the rate-limiting step of the mevalonate pathway [8]. In addition to their use in the treatment of lipid disorders, statins have been investigated for their anticarcinogenic effects in several models, including carcinomas of the colon and rectum, prostate, breast, lung, and skin [9,10]. The mechanism of apoptosis induction is well-known, and statin-induced suppression of angiogenesis via vascular endothelial growth factor and of tumor invasiveness and metastatic potential through interaction with adhesion molecules have shown anticancer effects [11]. The prevention of cardiovascular side effects of these statins, while in theory, as a safe drug is already widely used in clinical therapy with NSAIDs as interesting combination. In recent studies, NSAIDs and statins showed synergistic effects in combination with other therapeutics, such as peroxisome proliferator–activated receptor γ ligands, inhibitors of the epidermal growth factor receptor family, TRAIL receptor ligands, cisplatin, and doxorubicin [12]. We previously suggested that sulindac and simvastatin-induced reactive oxygen species (ROS) generation in A549 lung cancer cells causes ROS accumulation in mitochondria, triggering the release of apoptogenic molecules from the mitochondria to the cytosol, leading to caspase activation and cell death [13]. However, studies of the mechanism by which NSAIDs and statins combine to induce apoptosis and inhibit the growth of lung cancer cells are insufficient. Furthermore, the mechanism underlying deregulation of survivin by NSAIDs and statins in human non-small cell lung cancer cells has not been elucidated. In this study, we demonstrated the synergistic interaction of sulindac and simvastatin and elucidated the mechanisms underlying this synergy.
Materials and Methods
1. Reagents
RPMI 1640, fetal bovine serum (FBS), and antibiotics were purchased from Gibco-BRL (Grand Island, NY). Sulindac, simvastatin, propidium iodide (PI), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), bicinchoninic acid, dimethyl sulfoxide (DMSO), N-acetylcysteine (NAC), and LY294002 were purchased from Sigma-Aldrich (St. Louis, MO). The following primary antibodies were used caspase-3, -8, -9, poly(ADP-ribose) polymerase (PARP; Santa Cruz Biotechnology, Santa Cruz, CA), serine/threonine protein kinase (AKT), phospho-AKT, survivin, X-linked inhibitor of apoptosis protein (XIAP), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, Beverly, MA). Anti-rabbit IgG-conjugated horseradish peroxidase (HRP) antibodies and an enhanced chemiluminescent (ECL) kit were purchased from Amersham Pharmacia Biotech (Buckinghamshire, UK).
2. Cell culture
Human lung cancer A549 cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
3. Cell viability assay
The effects of sulindac, simvastatin, and sulindac plus simvastatin treatment on cell viability were determined by MTT assay. Briefly, A549 cells were seeded at a density of 5×103 cells per well in 96-well plates in triplicate. After 24 hours, sulindac, simvastatin, and sulindac plus simvastatin were added at various concentrations, and the cells were incubated for 48 hours. To determine the cell viability, MTT was added to the cell suspension for 4 hours. After 3 washes with phosphate buffered saline (PBS; pH, 7.4) the insoluble formazan product was dissolved in DMSO. The optical density (OD) of each well was then measured using a microplate reader (Titertek Multiskan, Flow Laboratories, North Ryde, Australia) at 590 nm. The OD derived from formazan production in control cells was defined as 100% viability, and all other measurements were expressed as a percentage of the control cell value.
4. Detection of apoptosis
The degree of apoptosis was determined by measuring the percentage of cells showing hypodiploid DNA content upon flow cytometric analysis following staining of the cells with PI using the method described by Crissman and Steinkamp [14]. Cell cycle analysis was conducted using a FACScan equipped with the CellQuest software (Becton Dickinson, San Jose, CA).
5. Determination of ROS
To measure intracellular ROS, A549 cells were incubated in the dark with 10 μmol/L 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA; Molecular Probes, Eugene, OR) for 30 minutes. Cells were then washed, scraped gently, resuspended in PBS, and kept on ice for immediate analysis by FACScan flow cytometry using an argon laser (488 nm) for excitation. Green fluorescence due to intracellularly trapped DCF was collected on the FL1 channel on a log scale. Data were acquired and analyzed with the CellQuest program (Becton Dickinson).
To measure mitochondria-derived ROS, the mitochondriatargeted, O2 − sensitive, hydroethidine analog probe MitoSOX (M36008, Invitrogen Life Technologies, Carlsbad, CA) was used to determine relative O2 − levels. Briefly, cells were incubated with 5 μM MitoSOX for 10 minutes in RPMI-1640, washed twice with PBS, and then analyzed by FACScan flow cytometry.
6. Immunoblot analysis
Cells were harvested and lysed in protein extraction solution (PRO-PREP; Intron Biotechnology, Seoul, Korea) for 20 minutes on ice. Next, cell lysates were centrifuged at 15,000 rpm for 20 minutes at 4°C, after which the supernatant was mixed with a one-fifth volume of 5× sodium dodecyl sulfate (SDS) sample buffer, boiled for 5 minutes, and then separated by 10% SDS–polyacrylamide gel electrophoresis. Following electrophoresis, proteins were transferred to a membrane that had been blocked with TBS-T (25 mM Tris [pH, 7.6], 138 mM NaCl, and 0.05% Tween-20) containing 5% skim milk and incubated with primary antibodies at dilutions of 1:1,000-1:5,000. After a series of washes, the membranes were further incubated with secondary antibody (at 1:2,000-1:10,000) conjugated with HRP. The immunoreactive signal was detected using an ECL detection system (Amersham Bioscience, Buckinghamshire, UK).
7. Small interference RNA transfection
Transcriptional expression was specifically suppressed by the introduction of a 21-nucleotide duplex siRNA that targeted the nucleotides of the AKT or survivin mRNA coding sequence [15]. Briefly, cells (105 cells per well) were plated in 6-well plates and transiently transfected with 2 μg per well of AKT or survivin siRNA (Cell Signaling Technology) mixed with X-tremeGENE siRNA transfection reagent (Roche Applied Science, Penzberg, Germany) according to the manufacturer’s directions. Silencer Negative Scramble siRNA (Roche Applied Science) was introduced into the cells as a negative control using the same protocol.
8. Plasmid preparation overexpressing AKT or survivin
Human survivin cDNA was obtained by reverse transcription– PCR of RNA derived from A549 cells with the following primers based on GenBank accession No. U75285 (forward, 5'-GGGAATTCATGGGTGCCCCGACGTTGCC-3'; reverse 5'-CTCTCGAGTCAATCCATGGCAGCCAGCT-3'). PCR products were digested with EcoRI and XhoI and ligated into pcDNA3.1myc [16]. The myr-AKT1 cDNA was subcloned into the EcoRI and XhoI site of the pcDNA3.1myr-myc. For transient transfection, A549 cells were transfected with 2 μg Myr-AKT (catalogue No. 9008, Addgene, Cambridge, MA) or control vector using Lipofectamine 2000.
Results
1. Sulindac and simvastatin synergistically augment the apoptotic activity of A549 cells
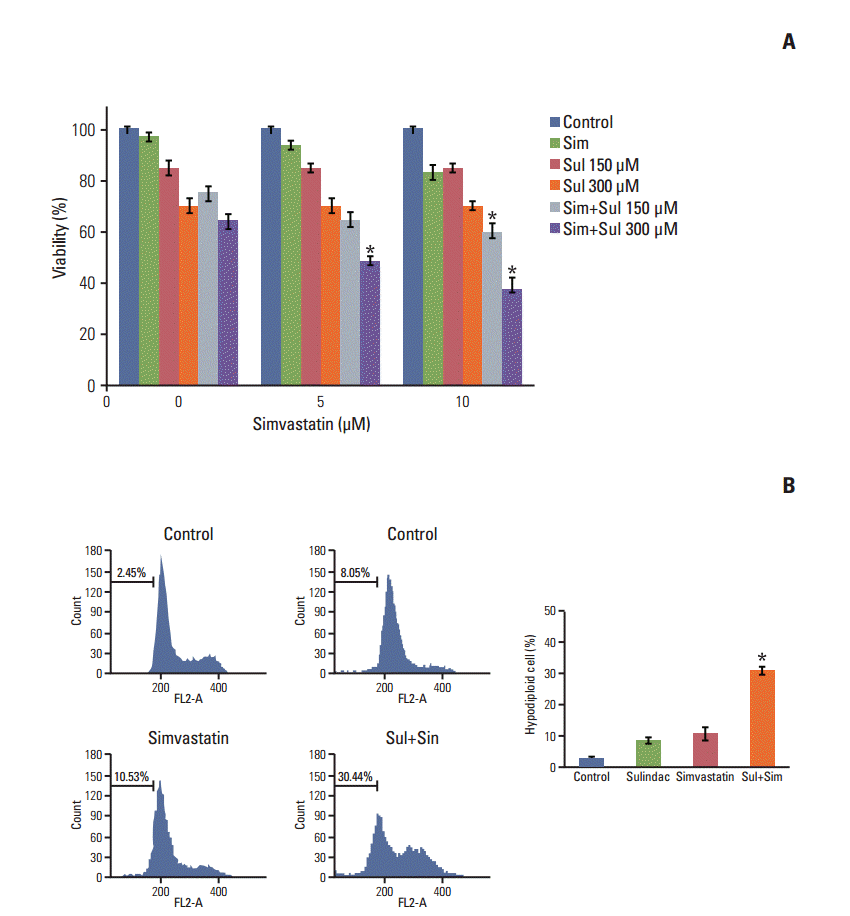
A549 cells were treated with sulindac and simvastatin, and their viability was measured by MTT assay. As shown in Fig. 1A, the combination of sulindac and simvastatin showed increased dose-dependent growth-inhibitory effects on A549 cells relative to treatment with sulindac or simvastatin alone. We next examined whether sulindac and simvastatin induced apoptosis in A549 cells. DNA fragmentation by apoptosis was considered the percentage of hypodiploid DNA content in A549 cells stained with PI measured by flow cytometry (Fig. 1B). The combination of sulindac and simvastatin caused a significantly greater increase in the accumulation of hypodiploid DNA content than either therapy alone. Taken together, these data indicate that combined treatment with sulindac and simvastatin triggers the apoptotic signaling pathway in A549 cells.
2. Combination of sulindac and simvastatin enhances intracellular ROS production
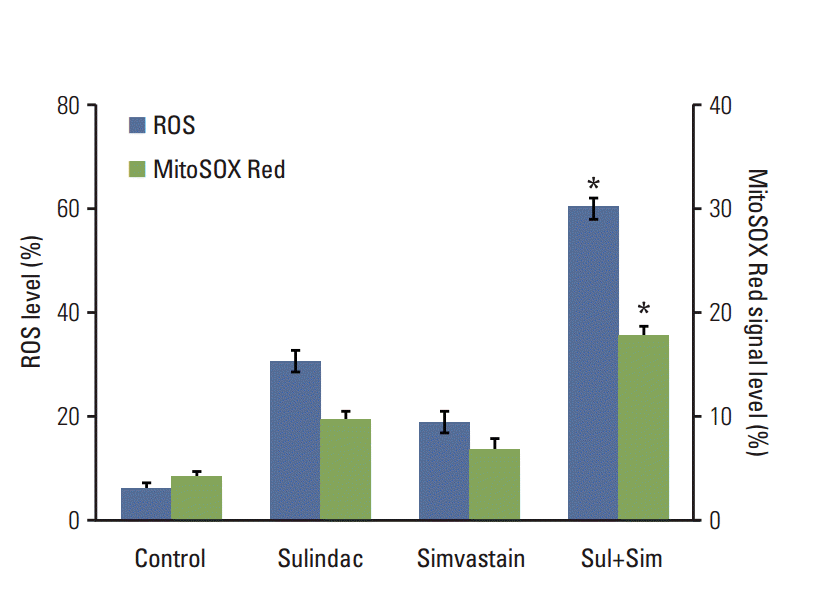
We next investigated the upstream regulatory mechanisms leading to the induction of apoptosis by combined treatment with sulindac and simvastatin. Intracellular ROS generation was assessed by flow cytometry using the total ROS marker H2DCFDA and the mitochondrial superoxide marker MitoSOX Red. Elevated ROS levels were detected 48 hours after combined treatment with sulindac and simvastatin (Fig. 2, blue bars), with marked ROS generation being observed in cells treated with both sulindac and simvastatin relative to cells treated with either compound alone. Flow cytometry measurement using MitoSOX revealed that intracellular O2 − levels increased significantly, which correlated well with the onset of total ROS production (Fig. 2, green bars). FACS analysis demonstrated an increase in the fluorescence intensity of MitoSOX Red in A549 cells. Specifically, the M2 levels were 9% and 6%, respectively, after 10 minutes of exposure to 300 μM sulindac or 5 μM simvastatin, but the M2 level was 18% in cells treated with both drugs.
3. Pretreatment with NAC prevents apoptosis and downregulates survivin by sulindac and simvastatin
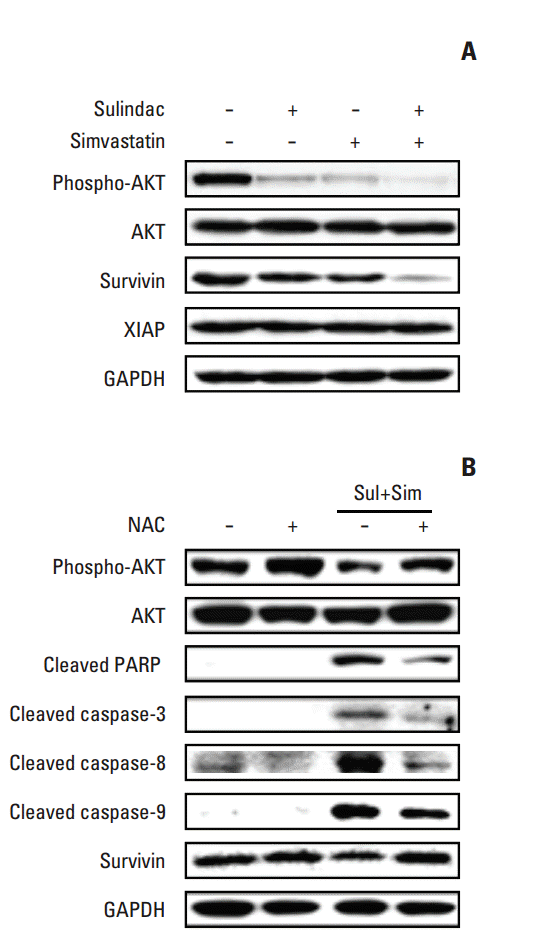
To identify the molecular mechanism underlying apoptosis induced by combined treatment with sulindac and simvastatin, we examined the expression of AKT and the inhibitor of apoptosis protein (IAP) family members survivin and XIAP by immunoblot analysis in A549 cells treated with sulindac and/or simvastatin for 24 hours. As shown in Fig. 3A, treatment of A549 cells with sulindac and simvastatin resulted in significantly lower survivin levels relative to treatment with either drug alone, but had no effect on the expression of XIAP. In addition, combined treatment with sulindac and simvastatin decreased phosphorylated AKT expression. To determine whether elevated ROS participates in apoptosis induced by the drug combination, cells were incubated with a free radical scavenger, NAC, before treatment. Western blot analysis of A549 cell lysates (Fig. 3B) showed that the combination of sulindac and simvastatin enhanced expression of the 85-kDa form of PARP and expression of caspase-3, -8, and -9, while pretreatment with NAC blocked this effect. Together, these findings indicate that ROS generation plays a primary role in apoptosis induced by sulindac and simvastatin.
4. Downregulation of survivin by sulindac and simvastatin is associated with suppression of the AKT signaling pathway
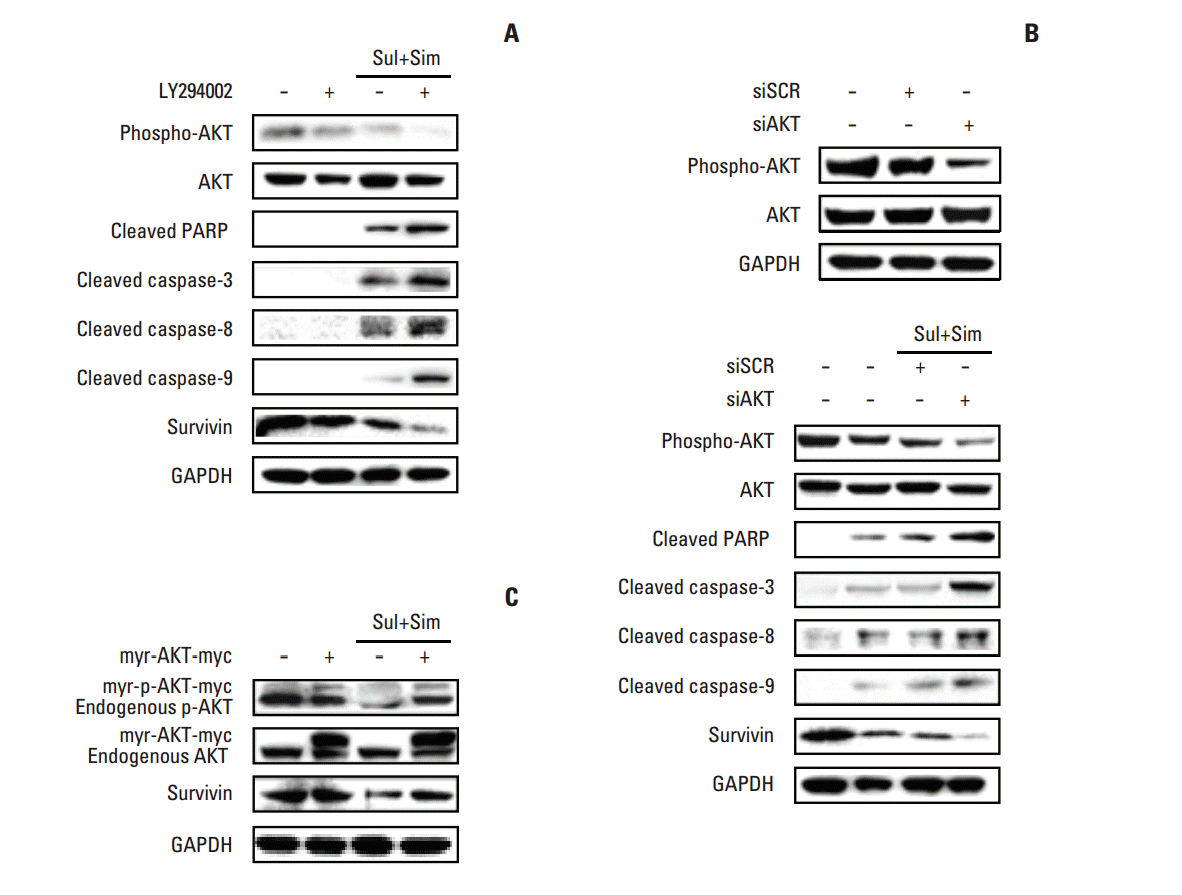
Because survivin downregulation involves the AKT signaling pathway, we tested the effects of the phosphatidylinositol 3-kinase (PI3K) inhibitor, LY294002, on A549 cells. To accomplish this, cells were pre-treated with or without LY294002 and then treated with sulindac and simvastatin, after which the cell lysates were assessed by Western blotting. As shown in Fig. 4A, pre-treatment with LY294002 decreased the activation of AKT, while treatment of cells with the combination of sulindac and simvastatin markedly decreased the levels of phospho-AKT. This effect was associated with significantly increased apoptosis, as evidenced by the cleavage of PARP and caspase-3, -8, and -9. The immunoreactive band of survivin was markedly decreased in cells treated with LY294002, as well as in cells treated with the combination of sulindac and simvastatin. We next knocked down AKT using siRNA. As shown in Fig. 4B, the introduction of AKT siRNA abrogated AKT protein expression 48 hours after transfection. Combined treatment with AKT siRNA, sulindac, and simvastatin resulted in greater cell death than combined treatment with scrambled siRNA, sulindac, and simvastatin. Next, we tested the effect of AKT activation by expressing a constitutively active form of AKT. As shown in Fig. 4C, treatment of A549 cells overexpressing AKT-myc with sulindac and simvastatin for 48 hours resulted in increased AKT and survivin relative to the control group. AKT overexpression markedly increased the p-AKT level, even in the presence of sulindac and simvastatin. The expression of survivin was also markedly elevated when compared with combination treatment. Collectively, these data suggest that the AKT pathway regulates survivin expression.
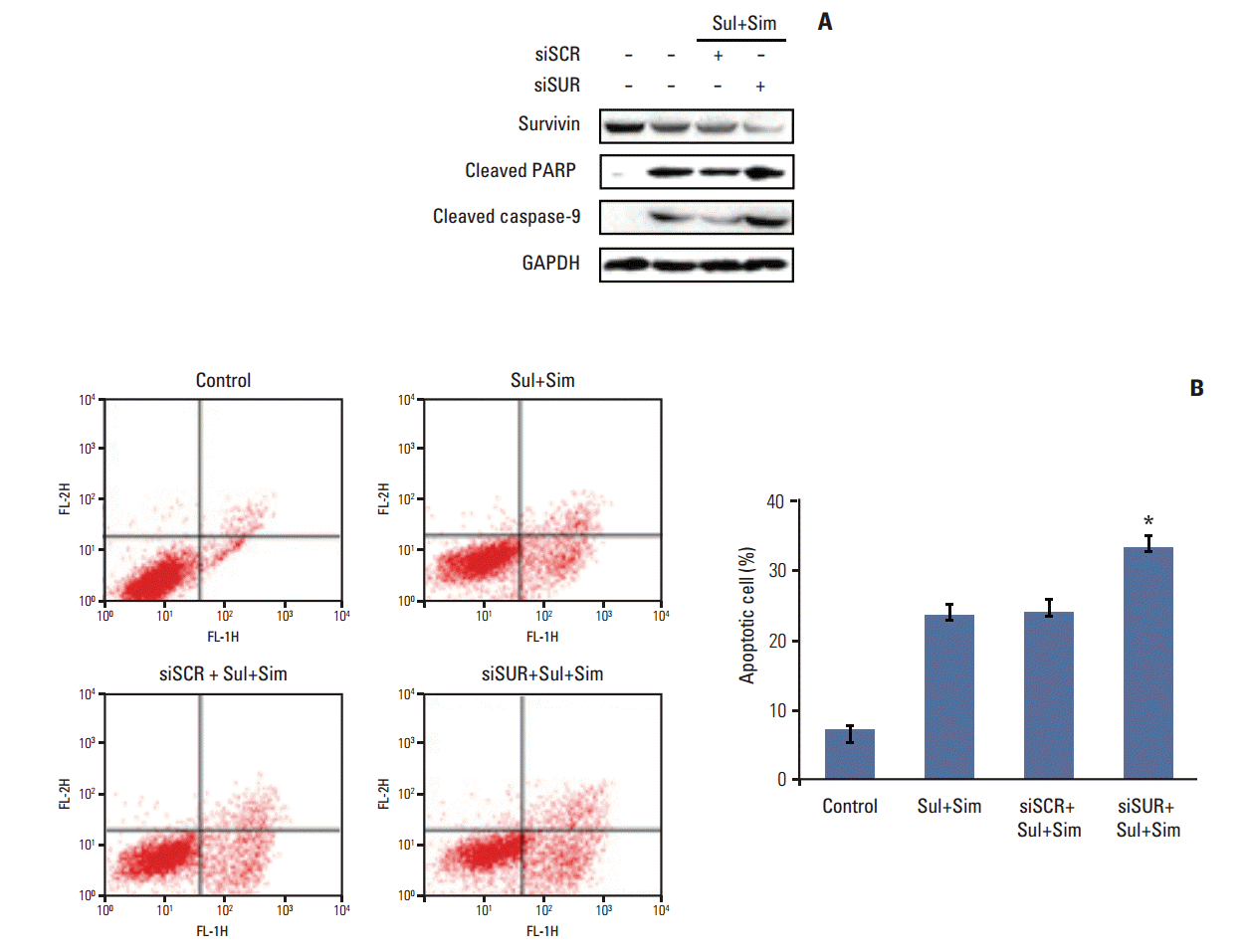
5. Downregulation of survivin augments apoptosis by sulindac and simvastatin
To determine the role of survivin in sulindac- and simvastatin-induced apoptosis in lung cancer, we knocked down survivin with siRNA. Introduction of survivin siRNA attenuated survivin protein expression 48 hours after transfection. No reduction in survivin protein was observed in cells transfected with the scrambled siRNA, which contained the same number of each nucleotide found in survivin siRNA. Cell death was higher in cells transfected with survivin siRNA than in those transfected with control siRNA. Moreover, the combination of sulindac, simvastatin, and survivin siRNA resulted in greater cell death than the combination of sulindac, simvastatin, and control siRNA (Fig. 5A). The number of annexin V–positive cells also increased in the survivin siRNA transfectants, and the combination of sulindac, simvastatin, and survivin siRNA increased the percentage of annexin V–positive cells to 32.4% (Fig. 5B). Taken together, these data indicate that the induction of apoptosis by sulindac and simvastatin is due, at least in part, to the downregulation of survivin.
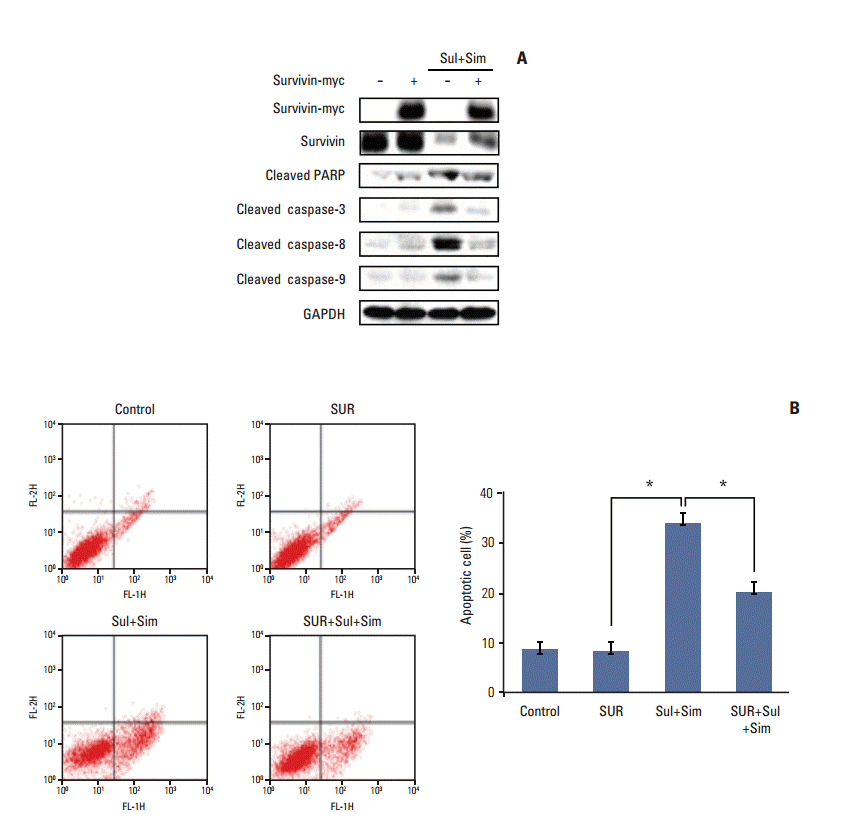
6. Upregulation of survivin attenuates apoptosis induced by sulindac and simvastatin
We next determined whether survivin upregulation played a role in apoptosis induced by sulindac and simvastatin. To accomplish this, we introduced a survivin gene expression vector, pcDNA3-myc-survivin, into A549 cells. The survivin gene transfectants expressed myc-tagged survivin protection in addition to endogenous survivin protection. The expression of cleaved PARP, caspase-3, -8, and -9 (Fig. 6A) was suppressed in survivin-overexpressing cells. Moreover, annexin V staining revealed that the number of apoptotic cells in control vector transfectants treated with sulindac and simvastatin for 48 hours was 34.2% higher than in untreated A549 cells. In contrast, the number of apoptotic cells in survivin gene transfectants treated with sulindac and simvastatin decreased by 19.5% (Fig. 6B). Together, these data indicate that the reduction of apoptosis by sulindac and simvastatin was due, at least in part, to the upregulation of survivin.
Discussion
In the present study, we showed that sulindac and simvastatin synergistically inhibit growth and induce apoptosis of A549 lung cancer cells, but that these effects were not observed when either drug was administered alone. The synergistic effect of sulindac and simvastatin on apoptosis has the potential to increase chemoprevention and treatment efficacy in lung cancer, which could enable a reduction in the dose of sulindac to minimize possible detrimental side effects. Additionally, simvastatin could be beneficial to cardiovascular health [10].
Apoptosis is one of the most vital pathways through which chemopreventive agents inhibit the overall growth of cancer cells; thus, it is important to determine whether the inhibition of cell proliferation and induction of apoptosis by chemopreventive agents are associated with the downregulation of antiapoptotic genes, such as survivin, which plays a critical role in cancer cell progression [17].
Many chemotherapeutic strategies have been designed to significantly increase cellular ROS levels with the goal of inducing irreparable tumor cell damage and death. Intracellular oxidative status is important for simvastatin sensitivity, and sulindac increases ROS levels more efficiently than selective COX-2 inhibitors [18]. A novel fluoroprobe, MitoSOX, was recently introduced for the selective detection of ROS in the mitochondria of live cells, and a quantitative method for the detection of mitochondrial ROS generation by flow cytometry was established [19]. Here, we adopted a staining technique using H2DCFDA and MitoSOX to concurrently detect intracellular and mitochondrial ROS generation in sulindac- and simvastatin-treated A549 cells. This simple method enables convenient observation of quantitative changes in ROS, both inside and outside the mitochondria. For novel therapeutic strategies it will be necessary to fine-tune intracellular ROS signaling to enable effective therapeutic gains.
Survivin, an IAP, regulates mitosis and inhibits apoptosis, primarily by suppressing the activity of caspase-3 [20]. Disruption of survivin-microtubule interactions results in loss of survivin anti-apoptotic function, increased caspase-3 activity, and cleavage of PARP. In addition, inhibition of the function of survivin induced apoptosis and increases in the responsiveness to chemotherapy or radiation therapy have been recognized as targets during the treatment of cancer [21]. There is evidence that the AKT-mediated survival pathway inhibits apoptosis by stimulating survivin synthesis in various cancer cell lines [22,23]. Therefore, we used siRNA-mediated knockdown of AKT or the PI3K inhibitor LY294002 in combination with sulindac and simvastatin treatment to determine whether the mechanism of enhanced apoptosis by combined treatment involves inhibition of the AKT pathway in lung cancer cells. It is known that the survivin signaling pathway to the insulin-like growth factor/mammalian target of rapamycin signaling pathway occurs via rapid changes in the mRNA transcription step [24]. Ras oncogene–related proteins or signal instruments and the activity of three transcription factors of the Wnt signaling pathway have been reported [25]. For future therapy, the upper signaling pathways that directly control survivin activity should be investigated.
Conclusion
Our study elucidated the involvement of AKT signaling– dependent downregulation of survivin in the anti-cancer effects of sulindac and simvastatin against lung cancer. These data provide a theoretical basis for future clinical use of sulindac and simvastatin in lung cancer chemoprevention and therapy and important information that will be useful for mechanistic and biomarker studies in clinical trials.
 XML Download
XML Download