

 PDF
PDF Citation
Citation Print
Print


Introduction
Cyclooxygenase (COX) is the key enzyme responsible for regulation of the conversion of arachidonic acid into prostaglandins and other bioactive lipids that help control both normal and aberrant cellular growth [1]. COX-1, which is the isoform constitutively expressed in many tissues, is believed to function as a housekeeping gene. Conversely, COX-2 is a pro-inflammatory factor that shows rapid up-regulation in response to stimuli such as mitogens, cytokines, growth factors, and tumor promoters [2]. COX-2 is important for modulation of apoptosis, angiogenesis, immune responses, and tumor invasion [2]. Increased COX-2 expression is frequently associated with various types of cancer including gastric carcinoma, non-small cell lung cancer (NSCLC), and esophageal carcinoma [3-5]. A preclinical study showed that COX-2 overexpression is associated with more aggressive tumor behavior and poorer prognosis among NSCLC patients [6]. Other investigations have shown that COX-2 overexpression promotes tumorigenesis, whereas nonsteroidal anti-inflammatory drugs and COX-2– specific inhibitors suppress tumorigenesis and tumor progression [7,8]. Thus, COX-2 inhibition might be important for the prevention of tumor formation and growth. Although many factors that enhance COX-2 expression have been identified, few that negatively modulate COX-2 production have been found.
Transforming growth factor β (TGF-β) is a multifunctional cytokine that functions as an important regulator of tissue homeostasis via induction of cell cycle arrest, differentiation, and apoptosis, as well as prevention of genomic stability [4,9,10]. A large number of experimental and clinical studies have demonstrated that the TGF-β signaling cascade can function as a potent tumor suppressor in a variety of cell types, including most epithelial cells [9]. However, it has also been shown that TGF-β signaling may play a role in enhancement of malignant transformation and tumor progression during late stage tumorigenesis given that TGF-β exerts stimulatory effects on COX-2 expression [3,4,11].
The expression of most genes can be regulated through multiple mechanisms including changes in mRNA stability. The stability of mRNA is regulated by a variety of signaling factors that primarily act on AU-rich elements (AREs), which are located in the 3' untranslated regions (UTRs) of short-lived mRNA molecules and serve as cis-acting destabilizing elements [12,13]. To date, a number of ARE-binding proteins, including tristetraprolin (TTP), have been shown to be encoded by immediate-early response genes induced by various stimuli [13,14]. Functional studies with knockout mice have shown that TTP acts as a tumor suppressor by regulating cellular signaling events that control transcription, mRNA turnover, and post-transcriptional regulation of ARE-containing genes. Furthermore, TTP is important for destabilizing tumor necrosis factor alpha mRNA by directly binding to the ARE located in the 3′-UTR and for regulation of COX-2 expression by binding to the COX-2 mRNA 3′-UTR in cancer cells [13,15].
Several reports have shown that TGF-β enhances COX-2 expression by inducing COX-2 mRNA expression or increasing mRNA stability in a variety of human cancers [3,4,16]. Based on these observations, it has been suggested that stimulation or enhancement of COX-2 expression by TGF-β may contribute to the tumor-promoting effects of TGF-β during the late stages of tumorigenesis. However, no studies have evaluated the effects of TGF-β on COX-2 expression in cancer cells, even though a previous investigation found that COX-2 mRNA levels are reduced in the presence of TGF-β in mouse calvarial bone cells [17]. In a previous study, A549 human lung cancer cells were found to express high levels of COX-2 [18]. Despite studies examining whether COX-2 silencing inhibits cell proliferation in A549 cells, no investigations have clearly demonstrated the anti-tumor response of TGF-β signaling related to COX-2 expression in A549 cells. Therefore, we conducted the present study to assess the ability of TGF-β to regulate COX-2 expression in A549 human lung cancer cells, which express high levels of COX-2 and are responsive to TGF-β tumor suppressive signals. Here, we report that TGF-β suppressed COX-2 expression by destabilizing COX-2 mRNA due to increased TTP levels in A549 cells.
Go to : 
Materials and Methods
1. Cell culture and TGF-β treatment
Human lung adenocarcinoma A549 cells authenticated using short tandem repeat analysis were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were stored in liquid nitrogen, passaged for less than 6 months before use in this study, and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and gentamicin (10 μg/mL) at 37°C under a 5% CO2 atmosphere. Human recombinant TGF-β1 (PeproTech, Rocky Hill, NJ) was rehydrated in 4 mM HCl with 1 mg/mL bovine serum albumin and used at a final concentration of 5 ng/mL for all experiments.
2. Western blot analysis
Cells were washed with ice-cold phosphate buffered saline and incubated in extraction buffer (20 mM Tris-Cl [pH 7.4], 100 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 5 mM MgCI2, 0.1 mM phenylmethylsulfonyl fluoride, 0.1 mM pepstatin A, 0.1 mM antipain, 0.1 mM chemostatin, 0.2 mM leupeptin, 10 μg aprotinin, 0.5 mg/mL soybean trypsin inhibitor, and 1 mM benzamidine) on ice for 15 minutes. Equal amounts of total cell protein (100 μg) were loaded onto 10% sodium dodecyl sulfate–polyacrylamide denaturing gels and transferred to nitrocellulose membranes. The blots were then probed with anti-human COX-2, anti-Smad2, and anti-Smad3 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were also incubated with antibody specific for α-tubulin (Sigma Chemical Co., St. Louis, MO) as a loading control. Antibody binding was detected using an ECL system (Amersham Pharmacia Biotech, Piscataway, NJ).
3. RNA extraction and Northern blot analysis
A TRI Reagent kit (Molecular Research Center, Cincinnati, OH) was used to isolate total RNA from the A549 cells. Total cellular RNA (20 μg) was separated on 1% agarose gels containing formaldehyde in MOPS (0.2 M 3-N-morpholino-propanesulfonic acid, 0.05 M sodium acetate, and 0.01 M EDTA). The mRNA was then transferred onto nylon membranes (Schleicher and Schuell, Keene, NH) by capillary blotting and crosslinked to the membranes using ultraviolet light. Finally, the blots were hybridized with COX-2 cDNA to evaluate RNA expression. Equal loading in lanes was confirmed using 18S RNA as an internal control.
4. Actinomycin D chase experiment (mRNA stability assay)
Cells were grown in media with TGF-β. Cells were incubated for the indicated times following the addition of 5 μg/mL actinomycin D (Sigma Chemical Co.). Total RNA was subsequently extracted and real-time polymerase chain reaction (PCR) was conducted to quantify the levels of COX-2 mRNA.
5. Reverse transcription PCR and real-time PCR
cDNA was synthesized from 1 μg of total RNA using 1 U of ImProm-II Reverse Transcriptase (Promega, Fitchburg, WI) and random hexamers. The TTP gene was amplified by PCR using the following primers as described by Ogawa et al. [19]: 5′-TCA TCC ACA ACC CTA GCG AA-3′ (sense) and 5′-GAT GCG ATT GAA GAT GGG GA-3′ (anti-sense). The COX-2 gene was amplified with 5′-ATC ACA GGC TTC CAT TGA CC-3′ (sense) and 5′-TAT CAT CTA GTC CGG AGC GG-3′ (anti-sense) primers. PCR was performed in 20 μL reactions by subjecting the samples to 94°C for 5 minutes followed by 30 cycles of 94°C for 30 seconds and 60°C for 30 seconds (for TTP) or 94°C for 30 seconds and 55°C for 30 seconds (for COX-2) followed by 72°C for 30 seconds. To estimate the efficiency of cDNA synthesis for each cell line, β-actin was used as an internal control. The PCR products were resolved in 1.5% SeaKem agarose gels, stained with ethidium bromide, and photographed under ultraviolet light.
Real-time reverse transcription PCR (RT-PCR) was performed using an iCycler Real-Time PCR Detection System (Bio-Rad, Hercules, CA). DNA melting curves were generated to monitor product specificities. mRNA was quantified according to fluorescence resonance energy transfer using a SYBR Green 490 fluorophore. PCR amplification and melting curves were analyzed using the iCycler Optical System Software ver. 3.0 (Bio-Rad,). A SYBR Green stock (1:2,000 dilution) was prepared in water. Primers were diluted to 10 pM in water. Real-time PCR was performed in a total volume of 20 μL using the following protocol: 5 minutes at 95°C followed by 40 cycles of 30 seconds at 95°C and 30 seconds at 60°C (for TTP) or 55°C (for COX-2). All samples were run in triplicate. RNA was quantified using the ΔΔCt method and normalized relative to that of glyceraldehyde 3-phosphate dehydrogenase as an internal control.
6. Transient transfection and luciferase assays
Cells were seeded at a density of 8×104/well in 12-well dishes and grown to 70%-80% confluence in complete growth media containing 10% FBS. Various COX-2 promoter reporter constructs generously provided by Drs. Hiroyasu Inoue and Tadashi Tanabe at the National Cardiovascular Center Research Institute, Japan (400 ng/well) and 100 ng of pSV-β galactosidase vector were used to co-transfect the cells with Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After 4 hours, complete medium was added and the cells were incubated for an additional 18 hours. After 24 hours of transfection, the cells were treated with or without TGF-β for 6 hours. Luciferase activity was measured using a TR717 microplate luminometer (Applied Biosystems, Foster City, CA) and a Bioluminescent Reporter Gene Assay System (Tropix, Inc., Bedford, MA) according to the manufacturer’s instructions. A pGL2 basic control vector without the insert was used as a negative control for the transfection experiments. The levels of luciferase activity were normalized to that of β-galactosidase.
7. Smad2, Smad3, and TTP knockdown using small interfering RNA (siRNA)
For siRNA knockdown, 80 nM of control, Smad2, or Smad3 oligomer were used for transfection in a 60-mm dish using Lipofectamine 2000 (Invitrogen) for 4 hours, after which the transfection medium was replaced with normal medium. The target sequences of the siRNA molecules specific for Smad2, Smad3, and TTP were as follows: siRNA-control (non-specific), 5′-TTC TCC GAA CGT GTC AGC T-3′; siRNA-TTP, 5′-GTT GTG GAT GAA GTG GCA GCG-3′. The siRNA molecules specific for Smad2 and Smad3 were synthetic siRNA duplexes of 21 nucleotides each that were guaranteed to silence the expression of human and mouse Smad2 and Smad3 (Cellogenetics, CLG-1107 for siSmad2 and CLG-1108 for siSmad3).
8. Statistic analysis
Results are representative of at least three independent experiments and are presented as the mean±standard deviation. Comparisons between groups were made using a Student’s paired t test. A p < 0.05 was considered to indicate statistical significance.
Go to :
Results
1. TGF-β blocks spontaneous induction of COX-2 expression in A549 cells
Due to the well-known effects of serum on COX-2 expression, we first evaluated the basal expression of COX-2 protein in the presence or absence of serum in A549 cells prior to assessing the effects of TGF-β on COX-2 expression. We demonstrated that COX-2 protein levels spontaneously increased in cells grown with or without serum, although COX-2 expression was more strongly augmented under serum-starvation conditions (Fig. 1A). Interestingly, spontaneous induction of COX-2 expression was inhibited upon TGF-β stimulation, regardless of the presence or absence of serum in the medium (Fig. 1A). Northern blot analysis showed that TGF-β reduced COX-2 mRNA levels in a time-dependent manner, with initial changes observed within 1.5 to 2 hours of TGF-β treatment (Fig. 1B). Moreover, COX-2 protein levels decreased in parallel with reduced COX-2 mRNA expression (Fig. 1C), suggesting that TGF-β down-regulates COX-2 protein expression in A549 cells by lowering COX-2 mRNA levels.
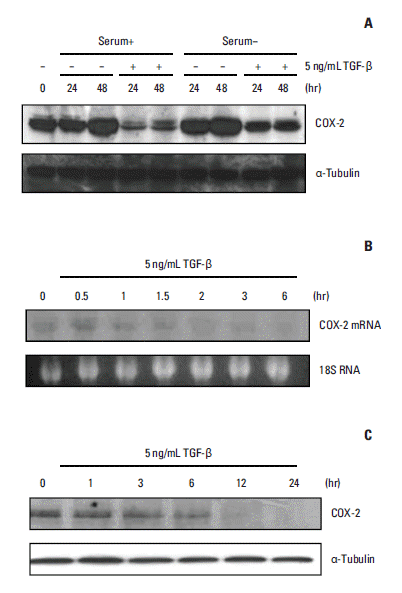 | Fig. 1.Expression of cyclooxygenase 2 (COX-2) mRNA and protein is suppressed by transforming growth factor β (TGF-β) treatment in A549 cells. (A) COX-2 protein levels were spontaneously elevated after cell seeding. This increase was more marked with serum starvation. TGF-β blocked the induction of COX-2 expression in the presence or absence of serum. A549 cells were incubated with or without 5 ng/mL TGF-β in the presence or absence of serum. Total cellular proteins were isolated at the indicated times, after which Western blotting was performed with anti–COX-2 or anti– α-tubulin antibodies. (B) COX-2 mRNA expression was down-regulated by TGF-β. The cells were treated with 5 ng/mL TGF-β for 1.5 to 2 hours, after which the total RNA was isolated at the indicated times and subjected to Northern blot analysis (20 μg per lane) to measure COX-2 mRNA expression. (C) COX-2 protein levels were down-regulated by TGF-β in A549 cells. The cells were treated with 5 ng/mL TGF-β. Total cellular proteins were isolated at the indicated times and analyzed by Western blotting as described above.
|
2. TGF-β does not affect COX-2 promoter activity in A549 cells
To determine whether the down-regulation of COX-2 mRNA expression by TGF-β is mediated at the transcription level, we investigated the effects of TGF-β on COX-2 promoter activity. To accomplish this, we used various COX-2 promoter-driven reporter constructs containing the regions spanning –1432 to +59 bp or –124 to +59 (Fig. 2A). Relative levels of COX-2 promoter activity were measured by a luciferase activity assay. None of the constructs assessed showed significantly altered expression in response to TGF-β (Fig. 2B), suggesting that regulation of COX-2 expression by TGF-β is not mediated by altered transcription rates in A549 cells.
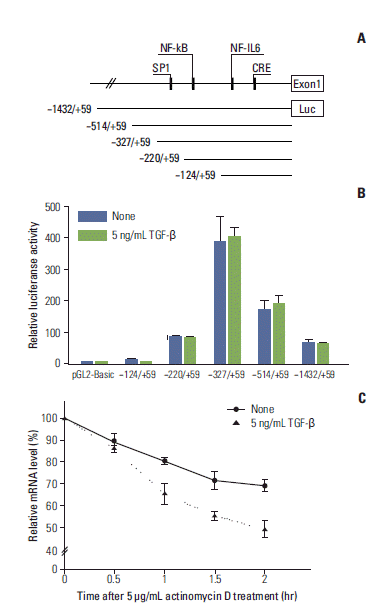 | Fig. 2.Transforming growth factor β (TGF-β) has no effect on cyclooxygenase 2 (COX-2) promoter activity, but affects COX-2 mRNA half-life. (A, B) Cells were transfected with a pGL2 basic control vector or the indicated reporter constructs, then treated with or without 5 ng/mL TGF-β for 6 hours. Levels of luciferase activity were measured as described in the Materials and Methods section. Data were normalized relative to β-galactosidase activity. No significant difference between the luciferase activities of TGF-β–treated and untreated cells was observed. (C) Cells were stimulated with TGF-β (5 ng/mL) for 1 hour, then treated with actinomycin D (5 μg/mL). The cells were harvested at the indicated times after actinomycin D was added. Total RNA was isolated, and real-time polymerase chain reaction was performed to measure the expression of COX-2 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The levels of COX-2 mRNA were normalized relative to GAPDH mRNA and presented as a percentage of that found in the untreated control. NF-kB, nuclear factor kB; IL-6, interleukin 6; CRE, cAMP response element.
|
3. COX-2 mRNA is post-transcriptionally regulated via destabilization by TGF-β
mRNA decay is critical for controlling the expression of many genes associated with inflammation and cancer via regulation of promoter activities. Since TGF-β did not affect COX-2 promoter activity in our system, we next examined the effects of TGF-β on COX-2 mRNA stability by an actinomycin D chase experiment. The rates of COX-2 mRNA decay were determined by quantifying the levels of COX-2 mRNA that remained in the presence of actinomycin D using real-time PCR. COX-2 mRNA decayed more rapidly in the presence of TGF-β (Fig. 2C). These results suggest that TGF-β triggers the down-regulation of COX-2 expression by decreasing mRNA stability rather than suppressing transcription.
4. COX-2 expression is down-regulated by TGF-β in a Smad3-dependent manner
Since TGF-β triggers various cellular responses through Smad2 and Smad3 [20], the primary intracellular mediators of TGF-β signaling, or a Smad-independent pathway, we determined whether the suppression of COX-2 expression by TGF-β is dependent on Smad2 and/or Smad3. To accomplish this, the effects of Smad2 and Smad3 knockdown on the TGF-β–associated inhibition of COX-2 expression were evaluated using RNA interference. No significant change in TGF-β–induced down-regulation of COX-2 expression was observed in the presence of Smad2-specific siRNA (Fig. 3). In contrast, Smad3-specific siRNA inhibited TGF-β–induced COX-2 down-regulation, resulting in COX-2 expression levels similar to basal levels (Fig. 3). These results suggest that the suppression of COX-2 expression by TGF-β is achieved via mRNA destabilization mediated by a Smad3-dependent pathway.
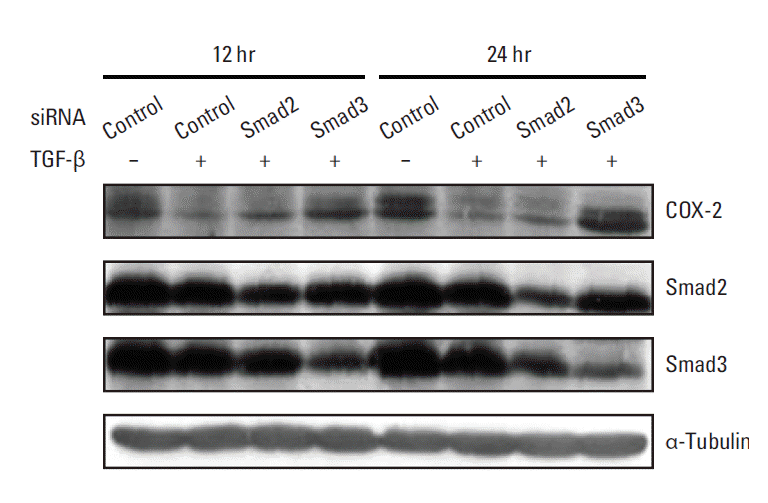 | Fig. 3.Smad3 knockdown restores the transforming growth factor β (TGF-β)–mediated down-regulation of cyclooxygenase 2 (COX-2) expression. A549 cells were transiently transfected with 80 nM of Smad2- or Smad3-specific siRNA. After transfection, the cells were treated with vehicle or 5 ng/mL of TGF-β for 12 or 24 hours, after which cell lysates were obtained for Western blot analysis. Total cell proteins (70 μg) were subjected to Western blotting and the membranes were probed with the indicated antibodies.
|
5. TTP expression is rapidly and transiently induced by TGF-β
TTP has recently been described as playing a pivotal role in the destabilization of COX-2 mRNA by directly binding to the 3′-UTR [13,15]. It was also reported that TGF-β transiently and rapidly induces TTP transcription by stimulating the binding of Smad3 to the TTP promoter in human T cells [19]. Based on these previous findings, we investigated whether TGF-β–induced COX-2 mRNA destabilization is mediated by TTP binding to the 3′-UTR of COX-2 mRNA. RT-PCR (Fig. 4A) and real-time PCR (Fig. 4B) analyses demonstrated that TTP expression was rapidly increased within 1 hour of TGF-β stimulation and abruptly decreased thereafter. These data suggest that TGF-β rapidly and transiently induced TTP expression in A549 cells, which is consistent with previous findings in other cell types [19].
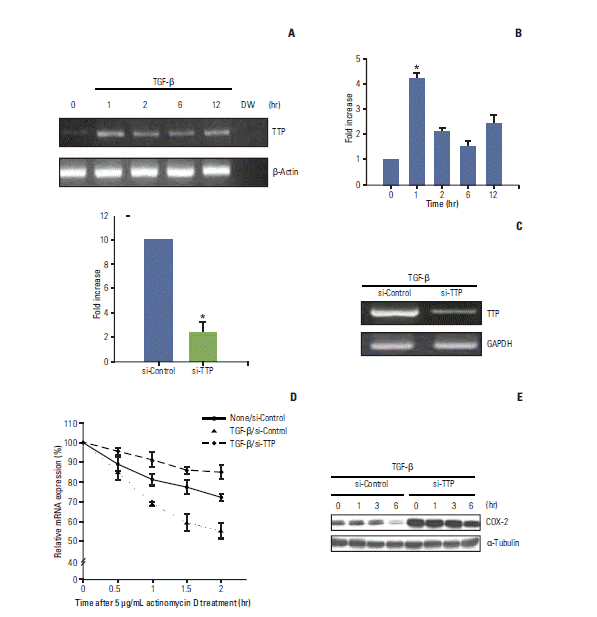 | Fig. 4.Tristetraprolin (TTP)-mediated cyclooxygenase 2 (COX-2) mRNA destabilization is induced by transforming growth factor β (TGF-β). (A, B) Total RNA was recovered from A549 cells treated with TGF-β (5 ng/mL) for different periods of time and Reverse transcribed. Reverse transcription–polymerase chain reaction (RT-PCR) and real-time PCR were then performed using primers specific for TTP, β-actin, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as described in the Materials and Methods section. The columns represent the mean of three independent experiments and are shown with error bars (±SE). *p < 0.001. (C) A549 cells were transfected with control or TTP-specific siRNA. After 24 hours of transfection, the cells were treated with TGF-β for 1 hour. RT-PCR and quantitative real-time PCR were used to measure the levels of TTP mRNA. Columns, the mean of three independent experiments; Bars, ±SE; *p < 0.001. (D) Cells transfected with control or TTP-specific siRNA were stimulated with or without TGF-β for 1 hour, after which actinomycin D (5 μg/mL) was added. Total RNA was isolated at different times, and COX-2 mRNA levels were quantified by real-time PCR. COX-2 mRNA expression was analyzed as described for Fig. 2C. (E) Cells transfected with control or TTP-specific siRNA were stimulated with TGF-β for the indicated times. Total cell proteins were then recovered and analyzed by Western blotting as described in the Materials and Methods.
|
6. TTP plays an essential role in TGF-β-mediated COX-2 mRNA destabilization
We next determined whether the rapid and transient increase in TTP expression helps reduce COX-2 mRNA stability. Using RNA interference, we eliminated TGF-β–induced expression of TTP and assessed subsequent changes in COX-2 mRNA stability. After confirming the successful knockdown of TTP by siRNA transfection and lack of response to TGF-β (Fig. 4C), we measured COX-2 mRNA stability. COX-2 mRNA stability (Fig. 4D) was unaffected by treatment with TGF-β in A549 cells transfected with TTP-specific siRNA. In contrast, COX-2 mRNA stability decreased in response to TGF-β in A549 cells transfected with the control siRNA. These results highlight the essential role of TTP in the TGF-β–induced destabilization of COX-2 mRNA. Interestingly, we found that the decay rate of COX-2 mRNA in TTP-depleted cells was greatly reduced and even lower than that observed in untreated cells (Fig. 4D). Thus, it is possible that TTP is also involved in regulation of the basal stability of COX-2 mRNA. Accordingly, the level of COX-2 protein was higher in TTP-depleted cells than control cells (Fig. 4E), even without TGF-β stimulation (Fig. 4E).
Taken together, these data are the first to indicate that the TGF-β–Smad3 pathway acts as a negative regulator of COX-2 expression via induction of TTP expression and subsequent TTP-dependent destabilization of COX-2 mRNA.
Go to :
Discussion
In the present study, we analyzed the molecular mechanisms underlying TGF-β–regulated expression of COX-2 in A549 human lung adenocarcinoma cells. In contrast to previous findings, we demonstrated that COX-2 expression was markedly suppressed in response to TGF-β [16,17]. In addition, TGF-β–induced inhibition was mediated by TTP-dependent COX-2 mRNA destabilization. Our data also indicated that TGF-β–activated Smad3 is a possible primary intracellular mediator responsible for the induction of TTP expression and subsequent reduction of COX-2 levels. Overall, we have provided the first evidence that TGF-β can negatively regulate COX-2 expression in A549 lung adenocarcinoma cells. Even though these cells cannot represent all types of human lung adenocarcinoma, our findings have helped us understand how TGF-β negatively affects tumor progression.
TGF-β is known to both promote and suppress tumor development in a cellular context–dependent manner [11]. These paradoxical effects of TGF-β on tumor growth occur because TGF-β functions as a tumor suppressor when the TGF-β signaling systems are intact and not masked by strong oncogenic signals. However, genetic and epigenetic alterations that impact the TGF-β systems and other pathways during tumorigenesis can compromise the TGF-β tumor suppressive activity. Under these conditions, TGF-β predominantly acts as an oncogenic factor that promotes tumor growth [3]. Given these opposing roles of TGF-β, positive regulation of COX-2 expression by TGF-β has been defined as increased COX-2 levels in response to TGF-β that enhances the oncogenic effects of TGF-β. The results from our study can be interpreted as evidence of the negative regulation of COX-2 expression by TGF-β that may promote TGF-β tumor suppressive activity. We conducted an MTT assay to evaluate the correlation between cell viability of A549 cells and TGF-β levels that down-regulate COX-2 expression. Finally, we examined a dose-dependent reduction of the cell viability accompanied with increasing levels of TGF-β (data not shown). This study also demonstrates that TGF-β can negatively regulate COX-2 expression through Smad3, a well-known mediator of TGF-β growth inhibitory signaling [11].
Gene expression is controlled both transcriptionally and post-transcriptionally. The regulation of mRNA stability is important for post-transcriptional modulation of gene expression [13]. The initiation and progression of tumorigenesis is characterized by the dysregulated expression of ARE-containing genes acting downstream of growth factor signaling [13]. For this reason, TTP can control the expression of several critical genes that are frequently up-regulated in cases of cancer via their ability to bind AREs and target mRNA for rapid degradation. Previous studies have shown that TTP binds to the ARE-rich 3′-UTR of COX-2 mRNA as a trans-acting factor, resulting in accelerated degradation of COX-2 mRNA [15]. Additionally, TTP inhibits the expression of COX-2 in human colon cancer [21]. Another report independently showed that TGF-β–activated Smad3 binds to the promoter region of the TTP gene, thereby transiently inducing TTP expression [19]. Consistent with these previous findings, the results of the present study demonstrated that TTP expression is transiently induced by TGF-β in A549 cells and plays a pivotal role in Smad3-dependent, TGF-β–mediated destabilization of COX-2 mRNA.
Even though the TGF-β pathway plays a role in the tumor suppressive response in human cancer, this factor also mediates tumor-promoting effects [22]. The tumor suppressor role is reinforced by alterations of the TGF-β signaling pathway members in human cancer cells that result in resistance to TGF-β–mediated effects associated with tumor progression. The tumor-promoting effect of TGF-β is demonstrated by elevated levels of this factor in patients with poorer prognosis during the advanced stages of cancer [23]. For these reasons, TGF-β and members of the TGF-β signaling pathway are being evaluated as prognostic or predictive markers for cancer patients.
Despite the prominent role of this pathway in tumor progression, targeting the TGF-β pathway for therapeutic purposes has been hampered by insufficient understanding of the regulation of this pathway in human cancer [24]. Additionally, increased TGF-β signaling could represent a chemoprevention strategy or a therapy for treating early-stage disease. This hypothesis is based on data suggesting that increased concentrations of serum TGF-β enhance the effects of the chemopreventive agents such as tamoxifen and retinoids [25]. Although increased TGF-β ligand activation may produce potential chemopreventive effects, the role of TGF-β activation in cancer treatment is still unclear.
Our findings further elucidate the mechanism of the TGF-β tumor suppressive role in cancer cells. These results may be helpful for assessing the potential for use of the TGF-β pathway for diagnostic, prognostic, or predictive purposes. Furthermore, targeting this pathway may serve as a novel strategy for treating human cancer.
Go to :
Conclusion
The present investigation was conducted to explore the relationship between TGF-β and TTP activation, as well as the subsequent effects on COX-2 mRNA stability in human lung adenocarcinoma cells. We found that TGF-β down-regulates COX-2 expression by Smad3/TTP-mediated COX-2 mRNA destabilization in A549 cells. These findings indicate that TGF-β functions as a negative regulator of COX-2 expression in a cellular context-dependent manner. To the best of our knowledge, this is the first study to provide evidence that TGF-β negatively regulates COX-2 expression in human cancer cells. In addition, we have identified an important new mechanism by which TGF-β controls the suppression of tumor development.
Go to :
 XML Download
XML Download