

 PDF
PDF Citation
Citation Print
Print


Introduction
Materials and Methods
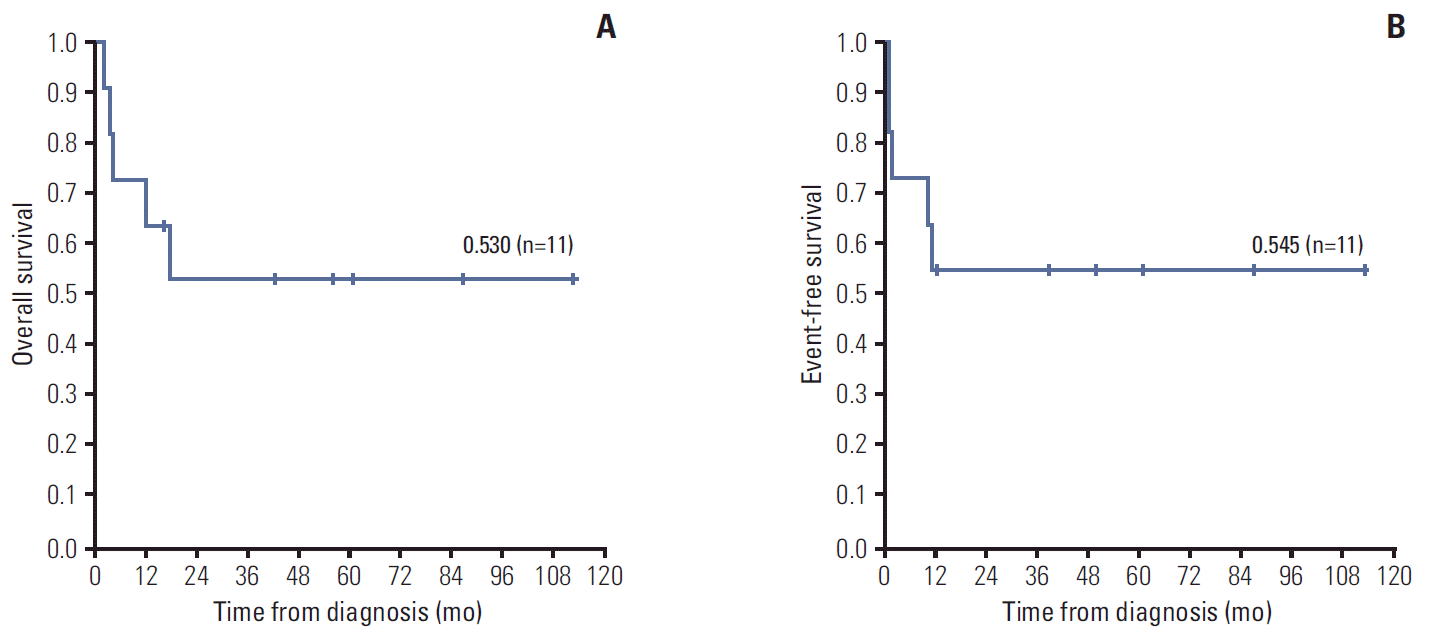
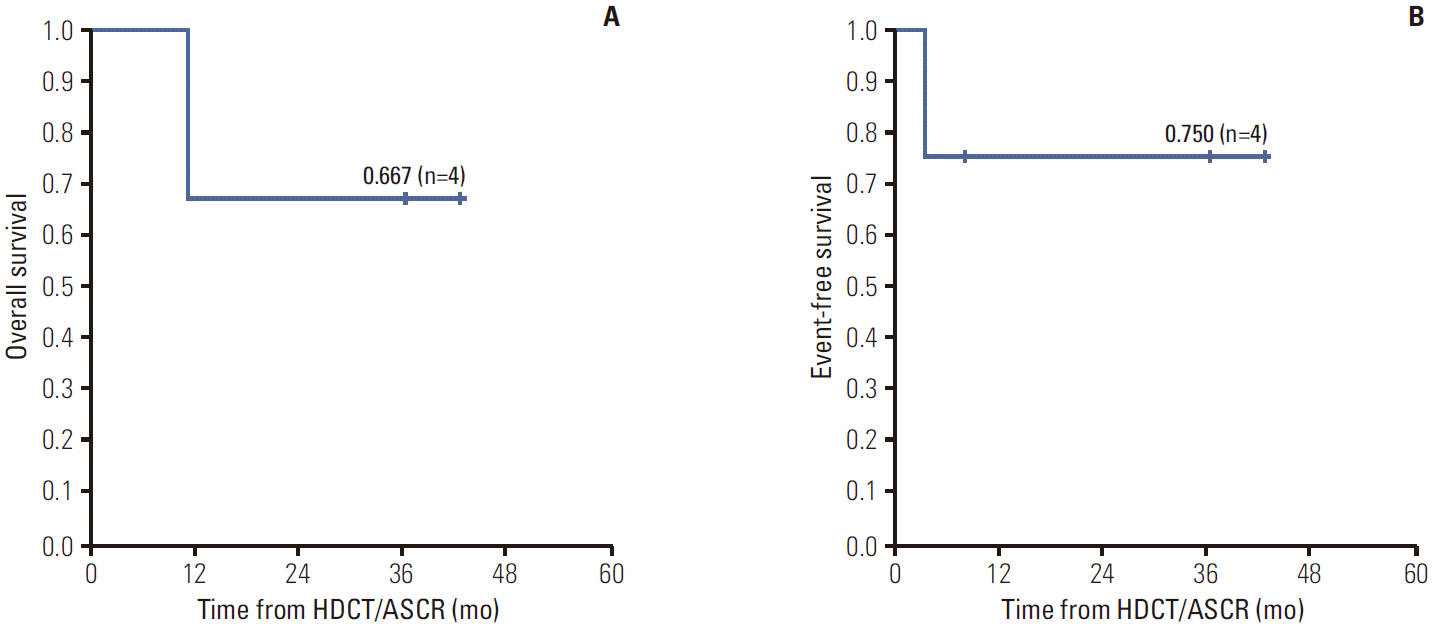
Results
1. Clinical characteristics
Table 1.
| Patient No. | Gender | Age at dx (mo) | Primary site | Presenting symptom | Metastatic site | SEER stage | Loss of INI1 | Chemotherapya) | HDCT/ASCR | Surgery (time from dx) | Therapeutic radiotherapy (time from dx) | Event (time from dx) | Outcome (time from dx) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 4 | Kidney, Rt | Gross hematuria | None | Localized | Loss | CbECy (wk24)→* | - | Upfront (at dx) | 10.5 Gy, 7 Fx (0.1 mo) | Relapse, brain (11.0 mo) | DOD (1 yr) |
| 2 | M | 5 | Kidney, Rt | Gross hematuria | None | Regional | Not evaluated | AV→AVD (wk3)→CbECy (wk24) | - | Delayed (0.4 mo) | 18.0 Gy, 12 Fx (1.7 mo) | None | NED (9 yr 4.3 mo) |
| 3 | M | 159 | Retrosternal area | Mass | None | Regional | No loss | CyE×2→CbE×2→(ICbE→VDCy)×7→(IE→VDCy)×2 | - | Delayed (4.1 mo) | None | None | NED (7 yr 2.4 mo) |
| 4 | F | 10 | Kidney, Rt | Gross hematuria | Lung | Distant | Loss | AV×2→†→ICbE→VDCy→CbECy (wk3) | - | Unresectable | None | Progression (0.9 mo) | DOD (4.5 mo) |
| 5 | F | 6 | Kidney, Lt | Abdominal mass | Lung, liver, bone (rid) | Distant | Loss | AV→CbECy (wk6)→†→IEpiP | - | Unresectable | None | Progression (1.7 mo) | DOD (3.8 mo) |
| 6 | M | 37 | Mass | Mass | Distant LN | Distant | Loss | (VDCy→IE)×3→IE×2→(VDCy→IE)×3→(VCy→IE)×2 | - | Delayed (4.4 mo) | 36.0 Gy, 20 Fx (6.5 mo) | None | NED (5 yr 0.5 mo) |
| 7 | M | 4 | Kidney, Lt | Gross hematuria | None | Regional | Loss | CbECy (wk24)→(VDCy→ICbE)×2→CyE→(VDCy→ICbE)×3→VDCy→MECb | Yes (MECb) | Upfront (at dx) | 21.6 Gy, 12 Fx (1 yr 2.3 mo) | None | NED (4 yr 7.8 mo) |
| 8 | M | 3 | Submental area | Mass | Back, periadrenal soft tissue | Distant | Loss | (VDCy→IE)×3→CyE→MECb→*→VPDCy/ITT→PEDCyV×3→ICbE×2 | Yes (MECb) | Upfront (at dx) | None | Relapse, brain/lung (10.2 mo) | DOD (1yr 5.8 mo) |
| 9 | M | 13 | Paraspinal | Mass | None | Localized | Loss | ICbE→VDCy→IE→VDCy→CyE→MECb | Yes (MECb) | Unresectable | 41.4 Gy, 23 Fx (7.8 mo) | None | NED (3 yr 6.2 mo) |
| 10 | F | 14 | Kidney, Rt | Fever, respiratory symptom | Lung distant LN | Distant | Loss | VDCy→†→ICbE1 | - | Delayed (2.1 mo) | None | Progression (1.0 mo) | DOD (2.3 mo) |
| 11 | F | 49 | Rt buttock | Rt toe pain | None | Localized | Loss | (VDCy→ICbE)×3→CyE→MECb | Yes (MECb) | Delayed (4.8 mo) | 32.4 Gy, 36 Fx (9.8 mo) | None | NED (1 yr 4.2 mo) |
dx, diagnosis; SEER stage, stage according to the Surveillance, Epidemiology, and End Results staging system; INI1, integrase interactor 1; HDCT/ASCR, high dose chemotherapy and autologous stem cell rescue; M, male; F, female; Rt, right; Lt, left; LN, lymph node; Gy, gray; Fx, fraction; DOD, died of disease; NED, no evidence of disease.
a)* relapse; †, progression; A, actinomycin D; Cb, carboplatin; Cy, cyclophosphamide; D, doxorubicin; E, etoposide; Epi, epirubicin; I, ifosfamide; ITT, intrathecal triple chemotherapy with methotrexate, hydrocortisone, and cytarabine; M, melphalan; P, cisplatin; V, vincristine; CbECy, intravenous (IV) carboplatin (16.7 mg/kg or 500 mg/m2 if body weight > 30 kg, d0-d1 on week 0, 3), IV etoposide (3 mg/kg or 100 mg/m2 if body weight > 30 kg, d0-d2 on week 0, 3, 9, 12, 18, 21) and IV cyclophosphamide (14.7 mg/kg or 440 mg/m2 if body weight > 30 kg, d0-d4 on week 6, 15, 24); AV, IV actinomycin D (0.015 mg/kg, d0-d2, d14-d16) and IV vincristine (d0, d7, d14, d21); AVD, IV actinomycin D (0.045 mg/kg, d0 of week 0), IV vincristine (1.5 mg/m2/wk, on week 1-3) and IV doxorubicin (22.5 mg/m2, d0 of week 3); CyE, IV cyclophosphamide (440 mg/m2, d0-d4, d21-d25) and IV etoposide (100 mg/m2, d0-d4, d21-d25); CbE, IV carboplatin (500 mg/m2, d0-d1, d21-d22) and IV etoposide (100 mg/m2, d0-d2, d21-d23); ICbE, IV ifosfamide (1,500 mg/m2, d0-d2), IV carboplatin (635 mg/m2, d2) and IV etoposide (100 mg/m2, d0-d2); VDCy, IV vincristine (1.5 mg/m2, d0, d7, d14), IV doxorubicin (30 mg/m2, d0-d1) and IV cyclophosphamide (1.8 g/m2, d0); IE, IV ifosfamide (2,400 mg/m2, d0-d4) and IV etoposide (100 mg/m2, d0-d4); IEpiP, IV ifosfamide (1,500 mg/m2, d0-d4), IV epirubicin (50 mg/m2, d0) and IV cisplatin (70 mg/m2, d0); VCy, IV vincristine (1.5 mg/m2, d0) and IV cyclophosphamide (1.8 g/m2, d0); MECb, high dose chemotherapy with IV melphalan (d-7 to d-6, 140/70 mg/m2), IV etoposide (d-8 to d-5, 200 mg/m2) and IV carboplatin (d-8 to d-5, 400 mg/m2); VPDCy/ITT, IV vincristine (2 mg/m2, d0, d7, d14), IV cisplatin (90 mg/m2, d0), IV doxorubicin (30 mg/m2, d1-d2), IV cyclophosphamide (300 mg/m2, d1-d3) and intrathecal triple with methotrexate (15 mg/m2), hydrocortisone (15 mg/m2) and cytarabine (30 mg/m2) on d0; PEDCyV, IV cisplatin (60 mg/m2, d0), IV etoposide (100 mg/m2, d2, d5), IV doxorubicin (30 mg/m2, d2), IV cyclophosphamide (30 mg/kg d3-d4) and vincristine (2 mg/m2, d0); ICbE1, IV ifosfamide (1,800 mg/m2, d0-d4), IV carboplatin (400 mg/m2, d0-d1) and IV etoposide (100mg/m2, d0-d4); CTx, chemotherapy.
![]()
 XML Download
XML Download