

 PDF
PDF Citation
Citation Print
Print


Introduction
Ovarian cancer is a leading cause of death in women worldwide, in part because symptoms often are not observed until the cancer has spread extensively. Thus, the prognosis for ovarian cancer is generally very poor and the mortality rate is high. Patients with ovarian cancer generally acquire resistance to chemotherapeutic agents and development of recurrent chemotherapy resistance is a common outcome of the cancer therapy, even in patients who have an initially positive clinical response to chemotherapy [1]. As such, developing a novel strategy against chemoresistance is a central issue.
Paclitaxel, isolated from the bark of Taxus brevifolia, is one of most potent chemotherapeutic agents against a wide spectrum of human cancers, particularly advanced ovarian and breast cancers [2]. Several studies have demonstrated that it induces apoptosis in various solid tumor cells. However, paclitaxel chemotherapy has had limited success because of dose-limiting toxicity and eventual drug resistance in patients. The molecular mechanisms of paclitaxel resistance are not well understood, although multifactorial mechanisms involving the activation of nuclear factor-κB (NF-κB), upregulation of cytoprotective pathways such as Akt and mitogen-activated protein kinase (MAPK), and alterations in β-tubulin isotypes have been suggested [3]. In addition, paclitaxel has been implicated in regulating targeted intracellular proteins that promote cell survival and alterations in the intrinsic apoptotic pathway [4].
Accumulating evidence suggests that activated NF-κB is critical in the development of drug resistance in cancer cells [5]. Therefore, agents that inhibit NF-κB function might be considered in combination with paclitaxel as an adjuvant approach for treatment of ovarian cancer.
Celecoxib, a class of non-steroidal anti-inflammatory drug, constitutes a potent and specific inhibitor of the human cyclooxygenase-2 (COX-2). COX-2 is constitutively overexpressed in many human premalignant, malignant, and metastatic epithelial tumors [6]. Upregulated expression of COX-2 may promote tumor growth through inhibition of apoptosis [7]. Moreover, COX-2 can be regulated by expression of vascular endothelial growth factor (VEGF), one of the most potent angiogenic factors known. In addition, one of the major positive regulators of COX-2 is NF-κB [8]. Therefore, the possibility of using COX-2 inhibitors as part of cancer therapy has gained interest. Mounting evidence suggests that celecoxib possesses potent antiproliferative, antiangiogenic, antimetastatic and proapoptotic properties in vitro as well as in vivo [7-9]. Celecoxib clinically promotes chemosensitivity and radiosensitivity of cancer cells. Celecoxib significantly prevents the development of chemoresistance in the breast cancer cell line MCF-7 [10], decreases NF-κB DNA binding, and inhibits cell growth in hepatoma cell lines [11]. However, though the anticancer effects of paclitaxel and celecoxib have been evaluated comprehensively in a variety of human cancers, the potential role of these compounds used in combination as an anticancer treatment has not been tested in ovarian cancer cells.
The present study investigated the potential role of celecoxib on paclitaxel-induced apoptosis in an ovarian cancer cell line. The results show that when celecoxib and paclitaxel are combined they have an additive or synergistic anticancer effect.
Go to : 
Materials and Methods
1. Cell culture
Human epithelial ovarian cancer cell line OVCAR-3 was cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin and 100 µg/mL of streptomycin. The cells were incubated in a humidified atmosphere containing 5% CO2 at 37℃.
2. Cell viability assay
Cell viability was measured using the Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Kumamoto, Japan). Cells were seeded in a 96-well flat-bottomed plate (2×103 cells in 100 µL per well). The cells were incubated overnight to allow for cell attachment and recovery and then exposed to paclitaxel (Bristol-Myers Squibb, New York, NY) and celecoxib (Pfizer Inc., New York, NY) for 6, 24, and 48 hours. Ten microliters of cell CCK-8 solution was added to each well, the cells were incubated for another two hours, and absorbance at 450 nm was measured with a microplate reader.
3. VEGF gene expression analysis by reverse transcriptase-polymerase chain reaction (PCR)
OVCAR-3 (2×106) cells were pretreated with celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM) for 6 hours. Total RNA was extracted using the Trizol reagent (Invitrogen, Grand Island, NY). One microgram of total RNA was converted to cDNA using oligo-dT as a primer and M-MLV reverse transcriptase (Invitrogen). PCR amplification proceeded as follows: 33 thermocycles at 94℃ for 30 seconds, 55℃ for 30 seconds, and 72℃ for 30 seconds, using oligonucleotides specific to human VEGF (forward: tga cag gga aga gga gga ga, reverse: tgg ttt caa tgg tgt gag ga) and human β-actin (forward: agg cca acc gcg aga aga tga cc, reverse: gaa gtc cag ggc gac gta gca c). Amplified fragments were analyzed using a 1% agarose gel and photographed over UV light. β-Actin was used as an internal control to compare data from different films.
4. Western blot analysis
Protein samples were lysed with 0.5% Nonidet P-40 and resuspended in complete lysis buffer. Proteins with 20 micrograms of cell lysate were directly separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Etten-Leur, The Netherlands). The membranes were blocked with 5% (w/v) non-fat milk in Tris buffered saline-Tween 20 (TBST) and incubated with primary antibodies (rabbit anti-p-NF-κB p65, anti-caspase-9, anti-poly ADP-ribose polymerase [PARP], anti-phospho-Akt and anti-γ-tubulin antibody; Cell Signaling, Beverly, MA) overnight at 4℃. The membrane was washed three times with TBST and then incubated with horseradish peroxidase (HRP)-linked anti-rabbit IgG secondary antibody for one hour at room temperature. After three 15-minute TBST washing steps, the protein was visualized on photography film (Pharmacia, Uppsala, Sweden) by enhanced chemiluminescence. The blots were reprobed with anti-γ-tubulin antibody for loading control.
5. Detection of NF-κB activity
Activation of the NF-κB p65 transcription factor subunits was determined using Trans AM kits according to manufacturer's instructions (Active Motif, Rixensard, Belgium). Nuclear extracts were prepared in hypotonic buffer and lysed with 0.5% Nonidet P-40. Concentration of the activated transcription factor was detected using an enzyme-linked immunosorbent assay (ELISA)-based method. The nuclear extract was incubated for 1 hour with a plate coated with an oligonucleotide that corresponded to a transcription factor consensus site. The wells were washed and incubated with an antibody against the indicated transcription factor subunit. An anti-rabbit IgG HRP conjugate was added, then the developing solution was added, and finally the colorimetric reaction was measured at 450 nm. The experiment was performed in triplicate.
6. Immunofluorescence
Cells were plated on eight-chamber slides (Nunc, Rochester, NY). After treatment, cells were rinsed in phosphate buffered saline (PBS) and fixed by incubation with 4% formaldehyde in PBS for 15 minutes at room temperature. After an additional wash with PBS, cells were permeabilized with PBS containing 0.25% Triton X-100 and then blocked in PBS containing 1% bovine serum albumin and 10% goat serum. The cells were incubated with polyclonal anti-p65 antibody in primary antibody dilution (1:200) buffer overnight at 4℃. After PBS washing, Alexa 488 (green) coupled secondary antibody (Invitrogen) was applied for 30 minutes at room temperature. Coverslips were mounted to slides using ProLong Gold antifade reagent containing DAPI and photographed using fluorescence microscopy.
7. Apoptosis assay
After exposure to celecoxib for 1 hour and incubation with paclitaxel for an additional 48 hours, cells were harvested by trypsinization. Cell density was adjusted to 1×106 cells per mL in PBS. Apoptosis was determined by staining with annexin V-FITC and 7-aminoactinomycin D (7-AAD) (BD PharMingen, San Diego, CA). Cells were analyzed by flow cytometry on a FACScan (Becton Dickinson, Mountain View, CA) using CellQuest software. In some experiments, apoptosis was also determined by ELISA using a DNA fragmentation kit (Roche Applied Science, Indianapolis, IN). Cells were seeded at a density of 2×104 cells per well in 96-well plates. After 24 hours, the medium was changed to a serum-free medium for at least 24 hours. To label DNA, the medium was replaced with 10% FBS-Dulbecco's modified Eagle medium and 10 µM 5-bromo-2'-deoxyuridine (BrdU) was added to each well and incubated for 24 hours. Cells were treated with celecoxib for 1 hour and then incubated with paclitaxel for an additional 48 hours. Cells were then lysed in 200 µL incubation buffer, and soluble DNA fragments were quantified using cellular DNA fragmentation ELISA according to the manufacturer's instructions. All experiments were performed in triplicate.
8. Measurement of caspase-3 activity
Caspase-3 activity was assayed in cellular extracts using the Caspase-3/CPP32 Colorimetric Assay kit (BioVision, Milpitas, CA) according to the manufacturer's instructions. Cell lysates were added to reaction mixtures containing 25 µM substrate DEVD-pNA. After incubation for 30 minutes, a spectrophotometer equipped with a 405 nm filter was used to detect caspase-3 activity. All experiments were performed in triplicate.
9. Statistical analysis
Statistical significance was determined using student's t-test. Null hypotheses of no difference between means were rejected if p-values were less than 0.05. Results are shown as mean±SD.
Go to :
Results
1. Celecoxib increased paclitaxel-induced growth inhibition of human ovarian cancer cell line OVCAR-3
The potential effect of celecoxib on paclitaxel-induced ovarian cancer cell death was tested using a cell viability assay (CCK-8 assay). A combination dose of 20 µM paclitaxel and 10 µM celecoxib significantly reduced cell viability (Fig. 1). These results indicate that celecoxib can dramatically enhance paclitaxel-induced ovarian cancer cell death by reducing cell viability in the human ovarian cancer cell line OVCAR-3.
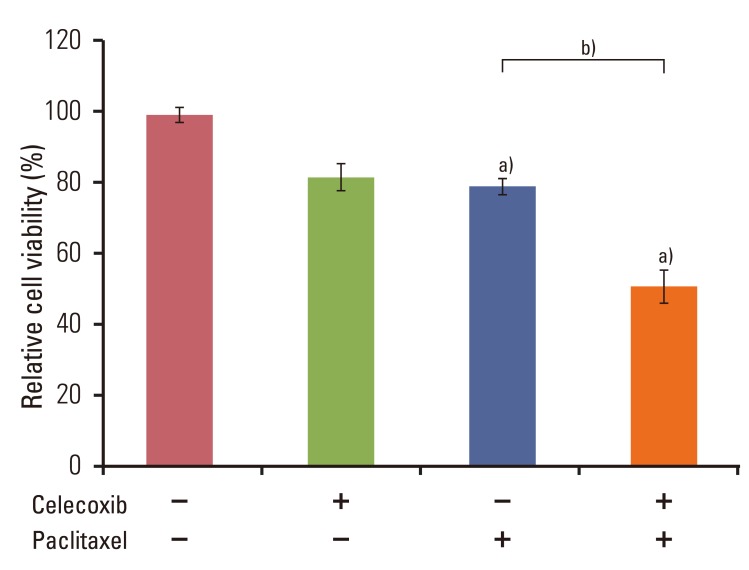 | Fig. 1Combined use of celecoxib and paclitaxel reduces cell viability of the human ovarian cancer cell line OVCAR-3. Cells were pretreated of celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM) for 6 hours. Cell viability was measured using the Cell Counting Kit-8 solution as described in the Materials and Methods section of this paper. Results are representative of three experiments. Data in the bar graph represent mean±SD. a)p<0.05 vs. control, b)p<0.05 vs. paclitaxel.
|
2. Celecoxib enhanced paclitaxel-induced apoptosis in OVCAR-3 cells
This study determined that celecoxib appears to increase apoptosis in cells that have been exposed to paclitaxel. The degree of apoptosis was assessed by measuring fluorescein isothiocyanate-conjugated Annexin V and 7-AAD double staining. Treatment with 20 µM of paclitaxel for 48 hours induced apoptosis (Fig. 2A). Celecoxib (10 µM) significantly increased apoptosis induced by paclitaxel in OVCAR-3 cells.
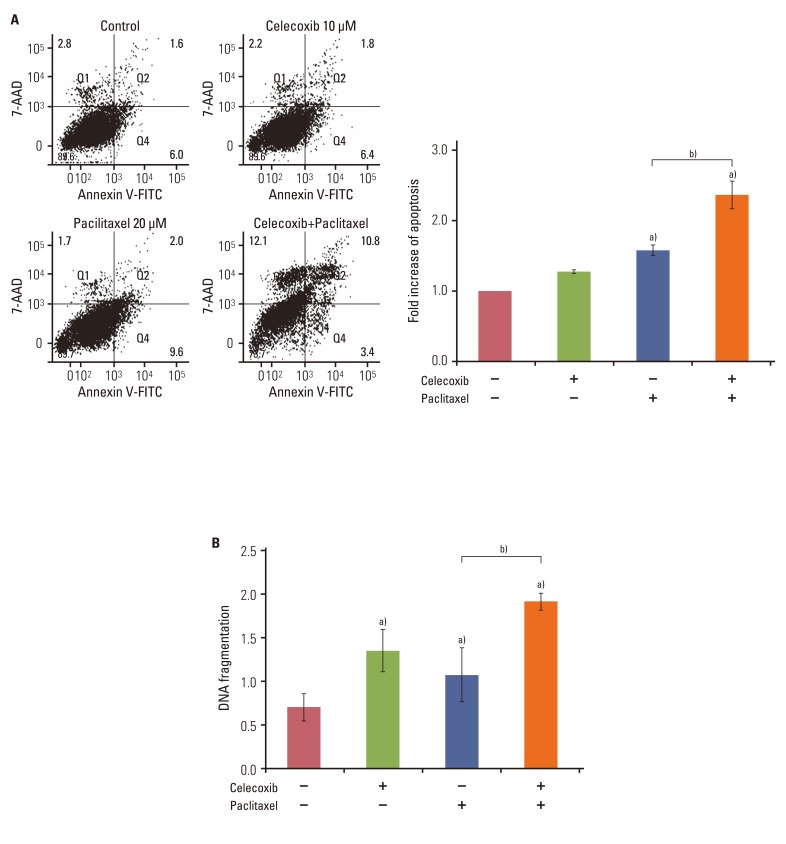 | Fig. 2Celecoxib promotes paclitaxel-induced apoptosis in OVCAR-3 cells. Cells (1×105 cells/well) were pretreated with or without celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM). (A) Apoptosis was assessed by Annexin V-FITC/7-aminoactinomycin D (7-AAD) staining after treating OVCAR-3 cells with paclitaxel for 48 hours, followed by fluorescence activated cell sorting analysis. Gate settings distinguish between living (bottom left), necrotic (top left), early apoptotic (bottom right), and late apoptotic (top right) cells. In the right panel, the levels of apoptosis are presented as the fold increase relative to the apoptosis of untreated OVCAR-3 cells. (B) Apoptosis was measured by cellular DNA fragmentation enzyme-linked immunosorbent assay. Results are representative of three experiments. Data are presented as mean±SD. a)p<0.05 vs. control, b)p<0.05 vs. paclitaxel treated group.
|
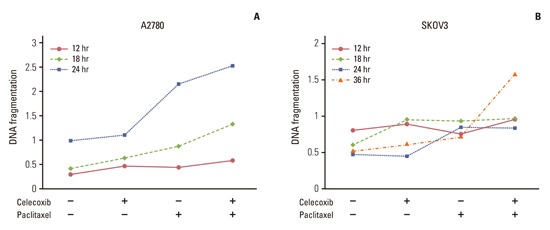
The degree of apoptosis was analyzed using an ELISA that measured the level of cellular DNA fragmentation. In accord with previous findings, paclitaxel induced DNA fragmentation in OVCAR-3 cells. Treatment with celecoxib enhanced DNA fragmentation induced by paclitaxel (Fig. 2B). To study the effect of celecoxib and paclitaxel on the other ovarian cancer cell lines A2780 and SKOV3, we evaluated apotosis by DNA fragmentation assay. When A2780 cells were treated with a combination of celecoxib and paclitaxel, there was significant DNA fragmentation in a time-dependent manner compared to paclitaxel treatment alone (Appendix 1A ). On the other hand, none of the combinations of celecoxib and paclitaxel induced any synergistic effect in SKOV3 cells (Appendix 1B ). Of note, in SKOV3 cells celecoxib increased DNA fragmentation induced by paclitaxel for 36 hours. These data suggest that celecoxib increased paclitaxel-induced apoptosis in ovarian cancer cells.
3. Celecoxib increased paclitaxel-induced caspase a activation and PARP cleavage
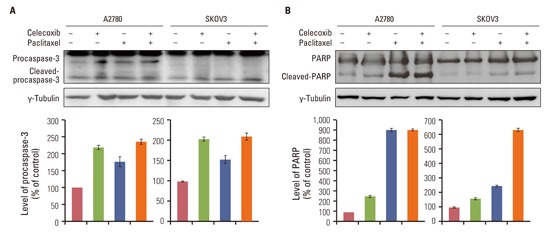
Cleaved caspase-9 and caspase-3 activity were evaluated by western blotting and colorimetric assay, respectively, to confirm that apoptosis occurred in response to a combination of celecoxib and paclitaxel. As shown in Fig. 3A, paclitaxel induced cleaved caspase-9 after 24 hours. Celecoxib significantly enhanced paclitaxel-induced activation of caspase-9 in OVCAR-3 cells. Moreover, combining celecoxib and paclitaxel significantly induced caspase-3 activation 24 hours after treatment in OVCAR-3 cells (Fig. 3B).
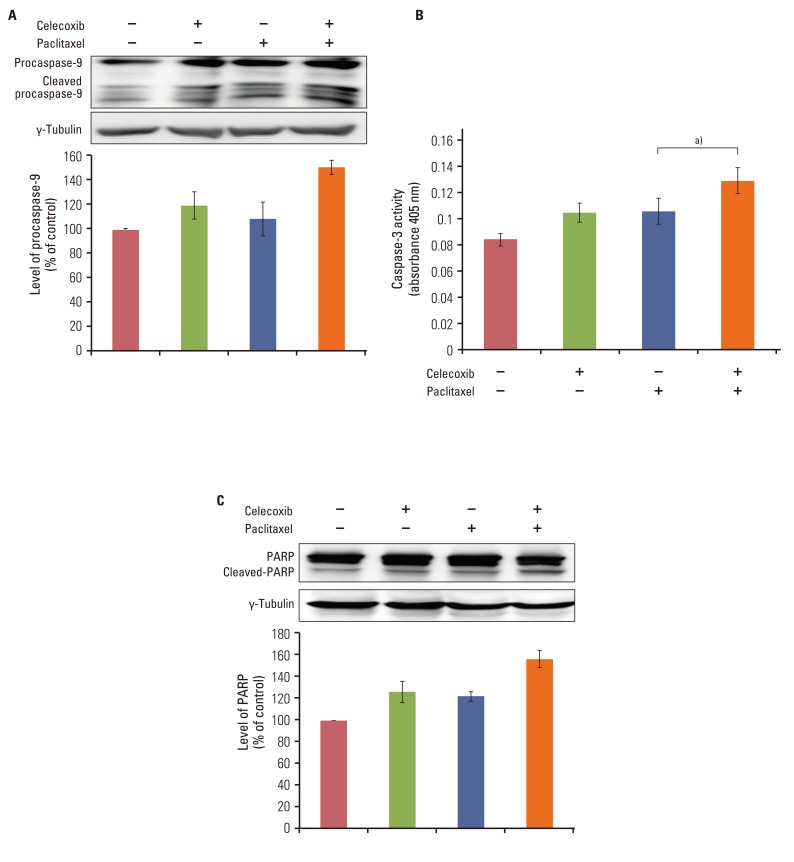 | Fig. 3Celecoxib enhances paclitaxel-induced caspase activation and poly ADP-ribose polymerase (PARP) cleavage. (A) Cell lysates from cells incubated in the presence or absence of celecoxib (10 µM) for 1 hour following stimulation with paclitaxel (20 µM) for 24 hours were subjected to western blotting with anti-procaspase-9 antibody. (B) Caspase-3 activity was measured by colorimetric assay. Data are presented as mean±SD. a)p<0.05 vs. paclitaxel treated group. (C) Cells were incubated with or without celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM). After 24 hours, cells were harvested and cleavage of PARP was analyzed by western blotting using anti-PARP antibody. The band intensities were quantitated.
|
Furthermore, PARPs are degraded by the caspase family as a consequence of apoptosis and have been used as markers of apoptosis. Cleavage of PARP was observed after treatment with a combination of paclitaxel and celecoxib (Fig. 3C). In sum, these results indicate that celecoxib increases paclitaxel-induced apoptosis in OVCAR-3 cells through a caspase-dependent pathway.
Celecoxib increased paclitaxel-induced activation of caspase-3 and PARP in A2780 cells (Appendix 2 ). The combination of celecoxib and paclitaxel led to a marked increase of caspase-3 activity and up-regulation of cleaved caspase-9 and cleaved PARP.
4. Celecoxib inhibited paclitaxel-induced NF-κB activation and VEGF mRNA expression
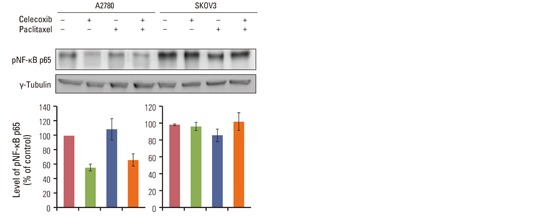
Paclitaxel activates NF-κB in several cell lines, and was also observed activating NF-κB in OVCAR-3 cells in this study (Fig. 4A and B). On the other hand, celecoxib decreased paclitaxel-induced NF-κB activation. Treatment with celecoxib alone decreased NF-κB activity. Immunofluorescence was performed against anti-p65 antibody to confirm that NF-κB is activated by paclitaxel (Fig. 4C). Pretreatment with celecoxib clearly down-regulated NF-κB activation induced by paclitaxel. Celecoxib inhibited paclitaxel-induced NF-κB activation in A2780 cells, as shown in the result from OVCAR-3 cells (Appendix 3). Interestingly, paclitaxel could not activate NF-κB in SKOV3 cells.
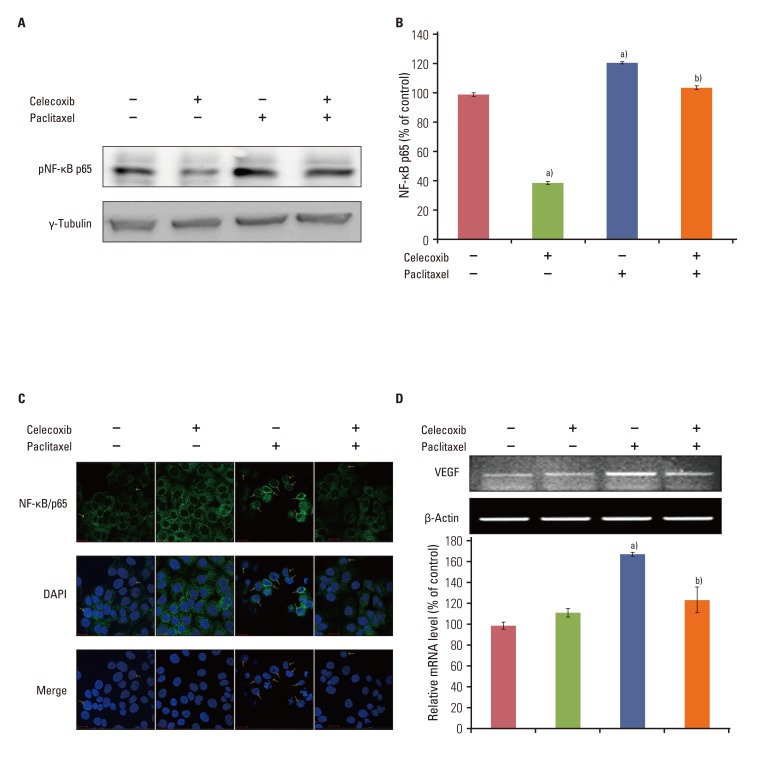 | Fig. 4Celecoxib reduces the activation of nuclear factor-κB (NF-κB) and vascular endothelial growth factor (VEGF) mRNA induced by paclitaxel. OVCAR-3 cells were pretreated of celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM) for 6 hours. The nuclear extract was collected. (A) Western blot analysis of pNF-κB p65. (B) The levels of active NF-κB family members were determined using the Trans AM NF-κB kit according to the manufacturer's instructions and reported as an absorbance level of the colorimetric substrate. (C) NF-κB/p65 localization was visualized using an anti-NF-κB/p65 primary antibody followed by an Alexa Fluor 488-conjugated detection antibody. (D) RNA was purified with Trizol as described in the Materials and Methods section of this paper. One microgram of total RNAs was subjected to reverse transcriptase-polymerase chain reaction for detection of VEGF mRNA expression. Results are representative of three experiments. Data in the bar graph represent mean±SD. a)p<0.05 vs. control, b)p<0.05 vs. paclitaxel.
|
VEGF mRNA expression was significantly induced by paclitaxel. Celecoxib decreased paclitaxel-induced VEGF mRNA expression 6 hours after treatment in OVCAR-3 cells (Fig. 4D). These data indicate that the combined use of celecoxib and paclitaxel in OVCAR-3 cells led to the inhibition of NF-κB activation and VEGF mRNA expression.
5. Celecoxib decreased paclitaxel-induced Akt activation in OVCAR-3 cells
We assessed potential molecular mechanisms supporting a combination treatment synergism of effectiveness by combining celecoxib and paclitaxel with emphasis on determining whether the combination treatment enhanced inhibition of the antiapoptotic signal transducers Akt. Celecoxib inhibits Akt phosphorylation. This study confirmed that paclitaxel induces activation of Akt after 24 hours (Fig. 5) and tested the effect of celecoxib on paclitaxel-induced Akt activation. Celecoxib pretreatment completely blocked the phospho-Akt band in the combination, indicating that Akt has a possible role in the synergistic effect of paclitaxel and celecoxib.
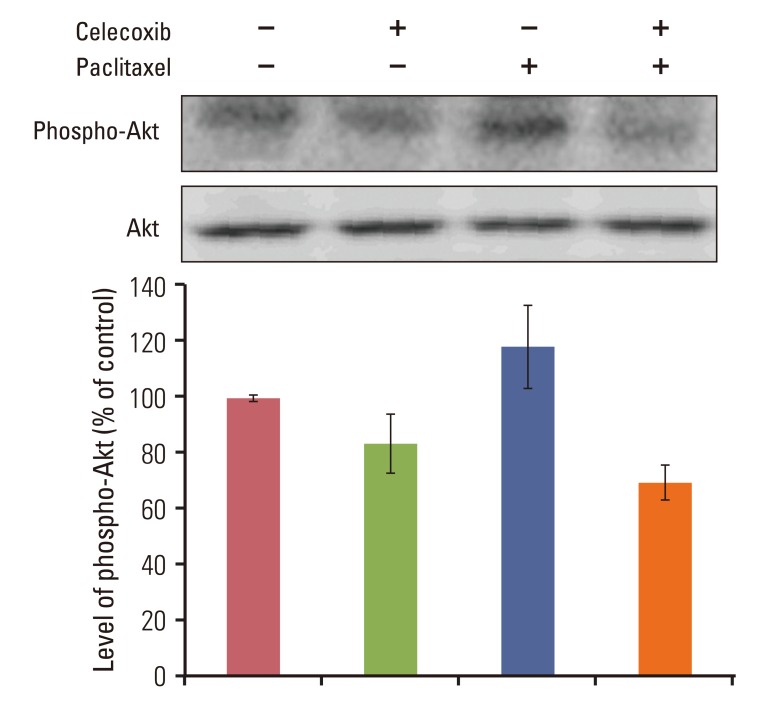 | Fig. 5Celecoxib reduces paclitaxel-induced Akt phosphorylation in OVCAR-3 cells. Cells were incubated with or without celecoxib (10 µM) for 1 hour before addition of paclitaxel (20 µM). After 24 hours, cells were harvested and Akt activation was analyzed by western blotting using anti-phospho-Akt antibody. The band intensities were quantitated.
|
Go to :
Discussion
This study investigated the synergistic anticancer effect of celecoxib and paclitaxel in the human ovarian cancer cell line OVCAR-3. Celecoxib significantly enhanced apoptosis induced by paclitaxel. This celecoxib-mediated enhancement of paclitaxel-induced apoptosis was accompanied by a significant increase in caspase activation and PARP cleavage. Additionally, celecoxib inhibited paclitaxel-induced NF-κB activation, VEGF expression and Akt phosphorylation in OVCAR-3 cells. These results suggest that combining celecoxib with paclitaxel contributes to chemopreventive activity by inducing apoptosis of ovarian cancer cells.
Most cytotoxic chemotherapeutic drugs cause apoptosis in cancer cells. However, certain tumor cells often escape apoptosis by overexpressing antiapoptotic proteins such as Bcl-2, NF-κB, and Akt [12-14]. Despite advances in surgery and chemotherapy, the survival rate of patients with ovarian cancer is still low because of drug resistance. The sensitivity of cells to chemotherapeutic agent-induced apoptosis seems to depend on the balance between proapoptotic and antiapoptotic signals. Paclitaxel is a microtubule-polymerizing agent that is widely used in the treatment of solid tumors [2]. In addition, it inhibits endothelial cell migration, invasion, capillary tube formation and proliferation in vitro and thereby leads to apoptosis in many cancers. A major disadvantage of paclitaxel is its dose-limiting toxicity and the development of drug resistance in patients. The development of novel strategies that produce outcomes similar to paclitaxel, but without chemoresistance, would greatly enhance the therapeutic effect of this drug. A recent study has shown that the specific COX-2 inhibitor NS398 inhibited cell growth and induced apoptosis in epithelial ovarian cancer cell lines [15]. Several studies have investigated the effects of COX-2 inhibitors in combination with various chemotherapeutic drugs. Celecoxib may be an effective potential chemopreventive agent. According to a recent report, combining celecoxib and paclitaxel produced disease stabilization in patients with metastatic melanoma [16]. Similarly, combining this COX inhibitor with either paclitaxel or cisplatin had a synergistic cytotoxic effect on human lung cancer cell lines [17]. This study showed that celecoxib significantly enhanced paclitaxel-induced apoptosis in OVCAR-3 and A2780 cells. The proapoptotic effect of combining celecoxib with paclitaxel was accompanied by a significant increase in caspase-3 activation and PARP cleavage.
NF-κB is a ubiquitous transcription factor that controls the expression of various genes associated with cell proliferation, angiogenesis, metastasis, the cell cycle, and antiapoptosis. In ovarian cancer, activation of NF-κB predicts poor prognosis and disease progression and confers resistance to cisplatin-induced apoptosis [18]. Increased NF-κB signaling by an inhibitor of NF-κB kinase B induces aggressiveness of ovarian cancer. One study found that increased activity of NF-κB reduces paclitaxel-induced apoptosis in ovarian cancer cells [19], while other studies have found that NF-κB mediates paclitaxel-induced apoptosis in ovarian cancer cells [20]. Thus the function of NF-κB in ovarian cancer depends on the experimental system and tumor types. NF-κB can function as a biphasic regulator, either suppressing or enhancing ovarian cancer growth through the regulation of MAPK and cellular apoptosis in parental or aggressive chemoresistant variant cell lines [21]. Moreover, the NF-κB pathway contributes to chemoresistance in ovarian cancer cell lines. In the present study, celecoxib increased paclitaxel-induced apoptosis and down-regulated NF-κB activation induced by paclitaxel in OVCAR-3 cells.
The current findings demonstrated down-regulation of paclitaxel-induced Akt activation by celecoxib in OVCAR-3 cells, and previous research has shown that the Akt survival cascade is related to chemotherapeutic drug sensitivity. Recent studies have discovered several agents that inhibit NF-κB and Akt, and these agents are currently in preclinical or clinical trials. Treatment with LY294002, a specific inhibitor of phosphoinositide 3-kinase, results in enhancement of paclitaxel-induced cytotoxicity through suppression of NF-κB activity [22]. Paclitaxel appears to activate NF-κB and Akt, which have critical roles in regulating cell survival, proliferation, invasion and metastasis [2,20]. On the other hand, celecoxib enhances apoptosis by interfering with various cell survival signaling pathways including NF-κB and Akt [23]. Given that Akt suppresses apoptosis by activating NF-κB, NF-κB may be the crucial intermediary step connecting Akt to the intrinsic susceptibility of cancer cells to chemotherapeutic agents [22]. Whether paclitaxel-induced up-regulation and celecoxib-induced down-regulation of Akt is regulated only through NF-κB is neither clear from these studies nor from ours. However, several studies have shown that paclitaxel directly activates the survival pathways such as Bcl-2, Akt, COX-2, or MAPK independently of NF-κB. This research attempted to determine the pathways contributing to the synergism of paclitaxel and celecoxib, and in doing so found that celecoxib downregulated paclitaxel-induced NF-κB and Akt activation. These results indicate that NF-κB and Akt are central to the synergism of paclitaxel and celecoxib.
COX-2 plays a significant role in tumor angiogenesis through up-regulation of VEGF production in several cancer cell lines [24]. VEGF appears contribute to angiogenesis in ovarian cancer. A relationship between COX-2 and angiogenesis has been noted, and hence COX-2 inhibitors may be effective antiangiogenic agents. Several studies have reported that selective COX-2 inhibitors suppress VEGF expression, angiogenesis and metastasis in vivo. One proposed mechanism of ovarian cancer prevention is a shared pathway dependent on the suppression of NF-κB activity, resulting in a downregulation of inflammatory mediators such as COX-2, VEGF, interleukin-8, and urokinase-type plasminogen activator [25]. Specific COX-2 inhibition using celecoxib suppressed serum VEGF concentrations in ovarian cancer. Additionally, VEGF suppresses apoptosis in lung and breast cancer cells and upregulates Bcl-2 levels in breast cancer cells, thus inhibiting apoptosis in both tumor cells and vascular endothelial cells. Celecoxib in combination with paclitaxel is an attractive antiangiogenic and anticancer treatment. These results confirm previous results indicating that celecoxib promotes cell apoptosis coupled with down-regulation of VEGF expression.
Go to :
Conclusion
Data from signal transduction studies and in vitro cell culture experiments strongly suggest that celecoxib enhances the antitumor activity of paclitaxel in ovarian cancer cells by modulating NF-κB activation and Akt inhibition. These findings indicate that treatment with paclitaxel in combination with celecoxib may be an effective clinical strategy for treating ovarian cancer, as combining the two agents enhances the antiangiogenic and antitumor effects of each agent alone.
Go to :
 XML Download
XML Download