

 PDF
PDF Citation
Citation Print
Print


Introduction
Tumor microenvironment plays an important role in tumor growth and metastasis. Tumor microenvironment consists of tumor cells and stromal cells including fibroblasts, endothelial cells, macrophages, dendritic cells, and lymphocytes, as well as these cells’ products such as extracellular matrix, cytokines, chemokines, growth factors, enzymes, and cellular metabolites. Macrophages can influence tumor growth, angiogenesis, invasion, and metastasis by expressing growth factors, cytokines, chemokines, and enzymes. The tumor-associated macrophages (TAMs) are a group of heterogeneous cells with a spectrum of diverse biological properties. The macrophages at the two ends of the spectrum are named M1 and M2 macrophages, mirroring the TH-1 and TH-2 nomenclature of T helper cells, respectively. Tumor necrosis factor α (TNFα), interferon-γ, lipopolysaccharides, and granulocyte-monocyte colony-stimulating factor are known to induce monocytes to differentiate into M1 macrophages. M1 macrophages express high levels of inducible nitric oxide synthase (iNOS), TNFα, interleukin (IL)-1β, IL-6, IL-12, IL-18, IL-23, CXC ligand 10, human leukocyte antigen-DR, and reactive oxygen and nitrogen intermediates. On the other hand, IL-4, IL-10, IL-13, IL-21, activin A, immune complexes, and glucocorticoids induce monocytes to become M2 macrophages [1]. M2 macrophages express high levels of IL-10, arginase I, IL-1 receptor antagonist, CC ligand 22, mannose receptor, galactose receptor, and CD163 antigen [1,2]. M1 macrophages inhibit tumor growth by producing effector molecules such as reactive oxygen intermediates, reactive nitrogen intermediates, and TNFα, whereas M2 macrophages promote tumor growth and metastasis by secretion of growth factors, vascular endothelial growth factor, matrix metalloproteinases, and immunosuppressive cytokines/chemokines [3]. The anti- or pro-tumor role of TAMs is determined by the balance between M1 and M2 macrophages [4]. We have previously reported that approximately 70% of TAMs are M2 macrophages and the remaining 30% are M1 macrophages in non-small cell lung cancers [5]. We have demonstrated that lung tumor tissues expressed significantly higher levels of IL-17 (also named IL-17A), cyclooxygenase-2 (COX-2), and prostaglandin E2 (PGE2) than normal lung tissues [6]. High levels of IL-17 in the lung cancer recruit monocytes /macrophages into the lung tumor microenvironment, and PGE2 induces them to differentiate into M2 macrophages [6]. However, it is not known if IL-17 also regulates the COX-2/PGE2 pathway in the cancer cells.
IL-17 binds to a heterodimer of IL-17 receptor A (IL-17RA) and IL-17 receptor C (IL-17RC). The activated receptor complex recruits nuclear factor-κB (NF-κB) activator 1 (Act1) through SEFIR (similar expression to fibroblast growth factor genes, IL-17 receptors and Toll–IL-1R) domains that exist in IL-17RA, IL-17RC, and Act1 proteins [7]. Act1 is an E3 ubiquitin ligase that activates tumor necrosis factor receptor-associated factor 6 (TRAF6) through lysine-63-linked ubiquitination [8]. Subsequently, the polyubiquitinated TRAF6 activates transforming growth factor-β-activated kinase 1 and IκB kinase complex, resulting in activation of NF-κB pathway that induces transcription of a variety of cytokines, chemokines and growth factors [7]. In addition, IL-17 activates the extracellular signal-regulated kinases 1 and 2 (ERK1/2) that stabilizes mRNAs of the IL-17 downstream target genes [9]. In this study, we found that IL-17 activated NF-κB and ERK1/2 pathways to up-regulate expression of COX-2 mRNA and protein in HeLa, A549, and Myc-CaP/CR cancer cell lines. Subsequently, the cancer cells secreted more PGE2 that acted on monocytes to promote M2 macrophage differentiation.
Go to : 
Materials and Methods
1. Cell cultures
Human cervical cancer HeLa cell line, human lung cancer A549 cell line, human THP-1 monocytes (from acute monocytic leukemia), and mouse RAW264.7 macrophages (from a mouse tumor induced by Abelson murine leukemia virus) were purchased from the American Type Culture Collection (Manassas, VA). Mouse castration-resistant prostate cancer cell line Myc-CaP/CR was a gift from Dr. Leigh Ellis and Dr. Roberto Pili (Roswell Park Cancer Institute, Buffalo, NY) [10]. HeLa, A549, Myc-CaP/CR, and RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s medium (Mediatech Inc., Manassas, VA) containing 10% fetal bovine serum (FBS; HyClone, Logan, UT) and 100 IU/mL penicillin/streptomycin. THP-1 cells were maintained in RPMI-1640 medium (HyClone) containing 10% FBS and 100 IU/mL penicillin/streptomycin. The cells were cultured in a 5% CO2 humidified incubator at 37°C.
2. Real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
After 15 hours of serum starvation, HeLa, A549, and Myc-CaP/CR cells were treated without or with 20 ng/mL recombinant IL-17A (R&D Systems Inc., Minneapolis, MN) for 2hours. The cells were collected in lysis buffer and homogenized with a 1-mL syringe connected to a 21-gauge needle. Total RNA was isolated according to the instructions of RNeasy Mini Kit (Qiagen, Valencia, CA) with on-membrane DNase I digestion to avoid genomic DNA contamination. cDNA was made from total RNA using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Human and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and COX-2 PCR primers were obtained from Eurofins MWG Operon (Huntsville, AL). Real-time quantitative PCR (qRT-PCR) was performed in triplicates with an iQ5 iCycler and iQ SYBR Green Supermix (Bio-Rad Laboratories) following the recommended protocols. Results were normalized to GAPDH levels using the formula ΔCt (Cycle threshold)=Ct of target gene–Ct of GAPDH. The mRNA level of the control group was used as the baseline, so ΔΔCt was calculated using the formula ΔΔCt=ΔCt of target gene–ΔCt of the baseline. The fold change of mRNA level was calculated as fold=2-ΔΔCt.
3. Induction of monocyte/macrophage differentiation
HeLa, A549, and Myc-CaP/CR cells were cultured in serum-free medium for 15 hours and then treated without or with 20 ng/mL IL-17A for 24 hours. The culture medium was centrifuged at 14,000 ×g for 10 minutes at 4°C. The supernatant from the untreated group was named conditional medium (CM) and the supernatant from the IL-17A-treated group was named IL-17A-induced CM. Approximately 2×106 THP-1 or RAW264.7 cells were cultured in serum-free medium in 60-mm dishes for 15 hours. The CM or IL-17Ainduced CM from human HeLa or A549 cells was used to treat mouse RAW264.7 cells. Conversely, the CM or IL-17Ainduced CM from mouse Myc-CaP/CR cells was used to treat human THP-1 cells. In this way, we could avoid crosscontamination of cancer cell mRNAs in qRT-PCR analysis of macrophage mRNAs because the PCR primers were speciesspecific. In addition, the control group was treated with serum-free medium without exposure to any cells and another group was treated with IL-17A in serum-free medium to assess the direct effects of IL-17A. After 3 hours of treatment, total RNA was isolated and qRT-PCR analysis was performed as described above. Human and mouse iNOS, TNFα, IL-10, arginase I, and GAPDH PCR primers were obtained from Eurofins MWG Operon.
4. Western blot analysis
To assess the effects of IL-17 on COX-2 protein expression, 2×106 cells were cultured in serum-free medium in 60-mm dishes for 15 hours. Then, the cells were treated without or with recombinant IL-17A (20 ng/mL) for 9, 12, and 24 hours. To assess the effects of IL-17 on NF-κB and ERK1/2 pathways, 2×106 cells were cultured in serum-free medium in 60- mm dishes for 15 hours. Then, the cells were treated without or with recombinant IL-17A (20 ng/mL) for 5, 15, 60, and 180 minutes. To assess the effects of pharmacologic inhibitors on IL-17-induced COX-2 protein expression, 2×106 cells were cultured in serum-free medium in 60-mm dishes for 15 hours. Then, the cells were treated without or with 100 μM NF-κB inhibitor pyrrolidine dithiocarbamate (PDTC; Sigma- Aldrich, St. Louis, MO) or 10 μM MEK inhibitor U0126 (Promega, Madison, WI), 30 minutes prior to addition of recombinant IL-17A (20 ng/mL) for 12 hours treatment. After the indicated time of treatment, proteins were extracted from the treated cells in RIPA lysis buffer (50 mM sodium fluoride, 0.5% Igepal CA-630 [NP-40], 10 mM sodium phosphate, 150 mM sodium chloride, 25 mM Tris pH 8.0, 1 mM phenylmethylsulfonyl fluoride, 2 mM ethylenediaminetetraacetic acid [EDTA], 1.2 mM sodium vanadate) supplemented with protease inhibitor cocktail (Sigma-Aldrich). Equal amount of proteins was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat dry milk in TBST buffer (25 mM Tris-HCl, 125 mM NaCl, 0.1% Tween 20) for 2 hours and probed with the indicated primary antibodies overnight and then IRDye 800CW- or IRDye 680RD-conjugated secondary antibodies (LI-COR Biosciences, Lincoln, NE) for 1 hour. The results were visualized by using an Odyssey Infrared Imager (LICOR Biosciences). For loading control, the membranes were stripped and probed for unphosphorylated proteins and/or GAPDH. The antibodies used are as follows: mouse anti- COX-2 antibodies were obtained from Cayman Chemical (Ann Arbor, MI, USA); goat anti-COX-2 and mouse anti-PERK1/2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti-ERK1/2, rabbit anti-P-IκBα, and mouse anti-IκBα antibodies were purchased from Cell Signaling Technology (Danvers, MA); mouse anti-GAPDH antibodies were ordered from Millipore Corporation (MAB374, Billerica, MA). Quantification of the Western blot signals was performed using the image analysis software of the Odyssey Infrared Imager system. The integrated density values of COX-2 signals were normalized by those of GAPDH. The ratio of COX-2/GAPDH indicates the relative level of COX-2 protein. The data represent the mean and standard deviation of three independent experiments.
5. Enzyme-linked immunosorbent assay of PGE2 levels
Approximately 2×106 cells of HeLa, A549, and Myc-CaP/CR cell lines were cultured in serum-free medium in 60-mm dishes for 15 hours. Then, the cells were treated without or with 20 ng/mL IL-17A for 24 hours. The culture medium was centrifuged at 14,000 ×g for 10 minutes at 4°C. The supernatant was collected and stored at −80°C until analysis. PGE2 levels were measured using a Prostaglandin E2 Enzyme Immunoassay Kit (Arbor Assays, Ann Arbor, MI) according to the manufacturer's instructions. Absorbance values were determined using a Bio-RAD Model 550 Microplate Reader. A standard curve was produced using a series of PGE2 concentrations from 31.25 to 1,000 pg/mL. The PGE2 level of each sample was obtained by plotting against the standard curve using CurveExpert 1.4 software (http://www.curveexpert.net). The data represent the mean and standard deviation of three independent experiments.
6. Statistical analysis
All experiments were repeated three times and the results represent mean±standard deviation of three independent experiments. Statistical analysis was made using two-tailed Student’s t-test. p < 0.05 was considered statistically significant.
Go to :
Results
1. IL-17 up-regulates expression of COX-2 mRNA and protein in cancer cell lines
We have previously shown that IL-17 up-regulates expression of chemokines and cytokines in human cancer cell lines such as HeLa, HEC-1-B, and SKOV3 [11]. In this study, we found that IL-17 significantly up-regulated COX-2 mRNA expression in HeLa, A549, and Myc-CaP/CR cancer cell lines (p < 0.05) (Fig. 1A). IL-17 also significantly up-regulated COX-2 protein expression in HeLa cells (p < 0.05) (Fig. 1B and C), A549 cells (p < 0.05) (Fig. 1D and E), and Myc-CaP/CR cells (p < 0.05) (Fig. 1F and G).
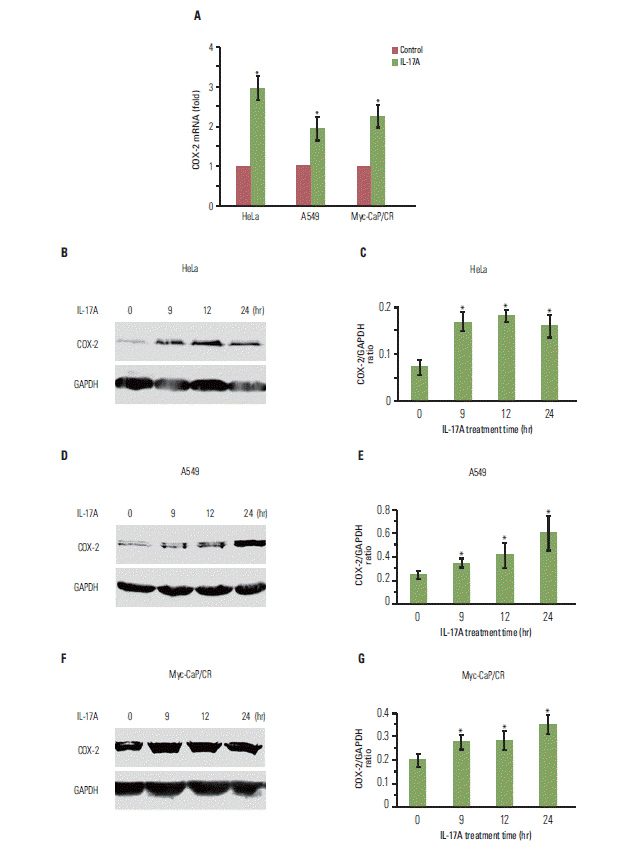 | Fig. 1.Interleukin (IL)-17 up-regulates expression of cyclooxygenase-2 (COX-2) mRNA and protein in cancer cells. (A) The cancer cells were treated without or with 20 ng/mL IL-17A for 2 hours; COX-2 mRNA levels were determined by real-time quantitative reverse transcriptase polymerase chain reaction. (B, D, F) The cancer cells were treated with 20 ng/mL IL-17A; COX-2 protein levels were determined by Western blot analysis. (C, E, G) Quantification of Western blot signals in three independent experiments. The ratio represents COX-2 signal divided by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) signal, where ratio=1 means that COX-2 signal is equal to GAPDH signal. Values are presented as the mean± standard deviation obtained from three independent experiments. *p < 0.05, compared to the control group.
|
2. IL-17 activates NF-κB and/or ERK1/2 pathways in cancer cell lines
We found that IL-17A increased phosphorylated IκBα (PIκBα) after 5, 60, and 180 minutes of IL-17A treatment in HeLa cells (Fig. 2A), suggesting that NF-κB pathway was activated in the IL-17A-treated cells. In contrast, P-ERK1/2 was not increased by IL-17A treatment (Fig. 2A), suggesting that ERK1/2 pathway was not activated by IL-17A in this cancer cell line. Similarly, we found that IL-17A increased P-IκBα after 15, 60, and 180 minutes of IL-17A treatment in A549 cells, however, P-ERK1/2 was not increased except a slight increase in P-ERK2 at 15 minutes (Fig. 2B), suggesting that NF-κB pathway was also the main signaling pathway that was activated in the IL-17A-treated A549 cells while ERK1/2 signaling pathway was only slightly activated in this cell line. In contrast, we found that IL-17A increased both P-IκBα and P-ERK1/2 in Myc-CaP/CR cells (Fig. 2C), suggesting that both NF-κB and ERK1/2 signaling pathways are activated in the IL-17A-treated Myc-CaP/CR cells. To determine if NF- κB and ERK1/2 signaling pathways mediate IL-17’s effects in regulation of COX-2 expression, we treated A549 and HeLa cells with an NF-κB inhibitor PDTC or MEK inhibitor U0126 prior to IL-17A treatment. We found that U0126 dramatically and significantly inhibited IL-17A-induced COX-2 protein expression in A549 cells (p < 0.05) (Fig. 3A and C), whereas PDTC only slightly but significantly inhibited IL-17A-induced COX-2 protein expression in A549 cells (p < 0.05) (Fig. 3A and C). In HeLa cells, both U0126 and PDTC only slightly inhibited IL-17A-induced COX-2 protein expression, however, the inhibition was statistically significant (p < 0.05) (Fig. 3B and D). In Myc-CaP/CR cells, neither U0126 nor PDTC inhibited IL-17A-induced COX-2 protein expression (data not shown). These findings suggest that IL-17 indeed acts through NF-κB and ERK1/2 signaling pathways in A549 and HeLa cells to regulate expression of downstream genes, which is consistent with our previous study [11].
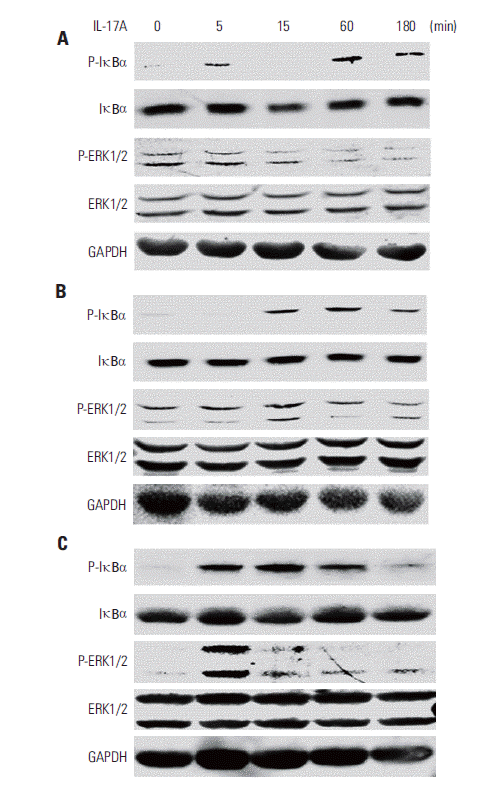 | Fig. 2.Interleukin (IL)-17 activates nuclear factor-κB and/or extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling pathways in cancer cells. HeLa cells (A), A549 cells (B), and Myc-CaP/CR cells (C) were treated without or with 20 ng/mL IL-17A; protein levels were determined by Western blot analysis. For protein loading control, the blots were stripped and reprobed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
|
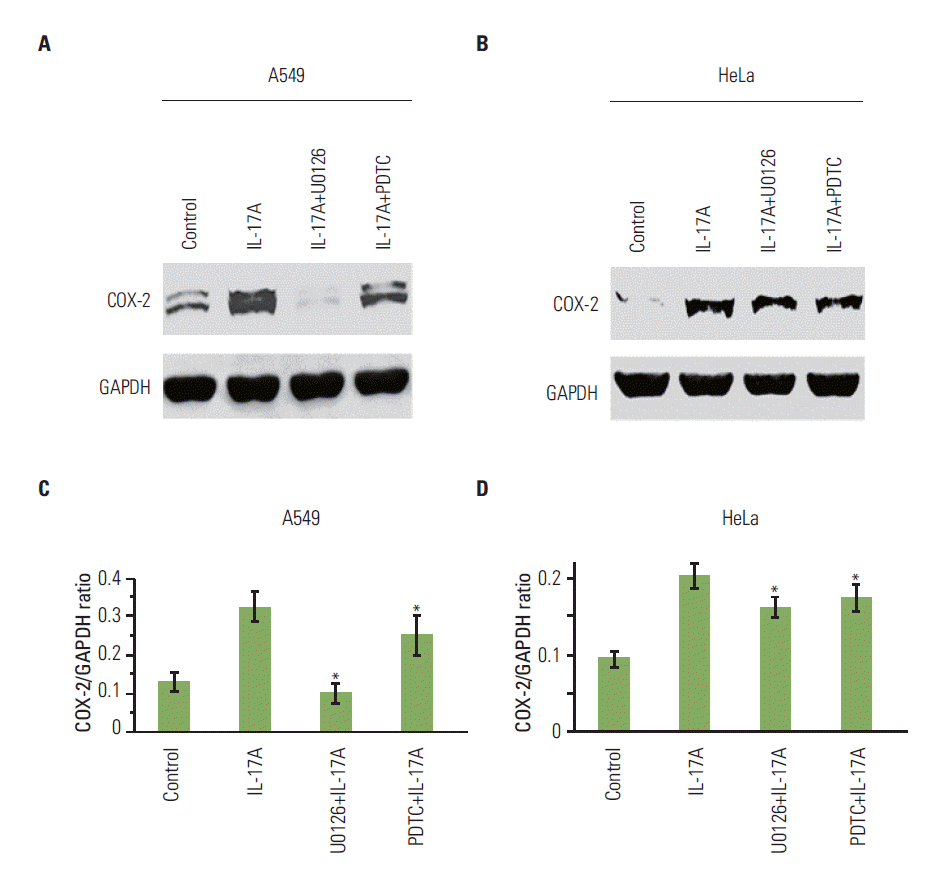 | Fig. 3.U0126 and pyrrolidine dithiocarbamate (PDTC) inhibit interleukin (IL)-17-induced cyclooxygenase-2 (COX-2) protein expression in cancer cells. (A, B) The cancer cells were treated without or with 100 μM nuclear factor-κB (NF-κB) inhibitor PDTC or 10 μM MEK inhibitor U0126 for 30 minutes prior to addition of recombinant IL-17A (20 ng/mL) for 12 hours treatment. COX-2 protein expression was determined by Western blot analysis. (C, D) Quantification of Western blot signals in three independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Values are presented as the mean±standard deviation obtained from three independent experiments. *p < 0.05, compared to the IL-17A treatment group.
|
3. IL-17 indirectly promotes M2 macrophage differentiation through induction of PGE2 synthesis
Since COX-2 is a critical enzyme for the production of PGE2 [12], we checked if IL-17 could induce PGE2 synthesis. We found that IL-17A significantly induced synthesis and secretion of PGE2 by HeLa, A549, and Myc-CaP/CR cancer cells (p < 0.05) (Fig. 4). Since we have previously demonstrated that PGE2 can induce RAW264.7 macrophages to express marker genes of M2 macrophages such as arginase I and IL-10 [6], we investigated if IL-17A-induced CM could show similar effects. We found that IL-17A-induced CM from HeLa cells significantly increased expression of iNOS, TNFα, IL-10, and arginase I, with the highest magnitude of induction seen in IL-10 (p < 0.01 or p < 0.05, compared to the IL-17A treatment group or CM treatment group, respectively) (Fig. 5A). The CM from HeLa cells also dramatically increased expression of iNOS, TNFα, and IL-10 (Fig. 5A), representing the effects of basal levels of PGE2 that was expressed in this cell line. In contrast, directly treating RAW264.7 macrophages with IL-17A did not dramatically increase expression of iNOS, TNFα, IL-10, or arginase I (Fig. 5A). Similar findings were obtained using the CM from A549 cells, again with the highest magnitude of induction seen in IL-10 (Fig. 5B). IL-17A-induced CM from Myc-CaP/CR cells significantly increased expression of TNFα and IL-10 in THP-1 monocytes (p < 0.01, compared to the IL-17A treatment group) (Fig. 5C), and the CM from Myc-CaP/CR cells also dramatically increased expression of TNFα and IL-10, with IL-10 at a higher level than TNFα (Fig. 5C). However, there was no statistical significance between the treatments with the CM and IL-17A-induced CM (Fig. 5C). Consistently, directly treating THP-1 monocytes with IL-17A did not increase expression of iNOS, TNFα, IL-10, or arginase I (Fig. 5C).
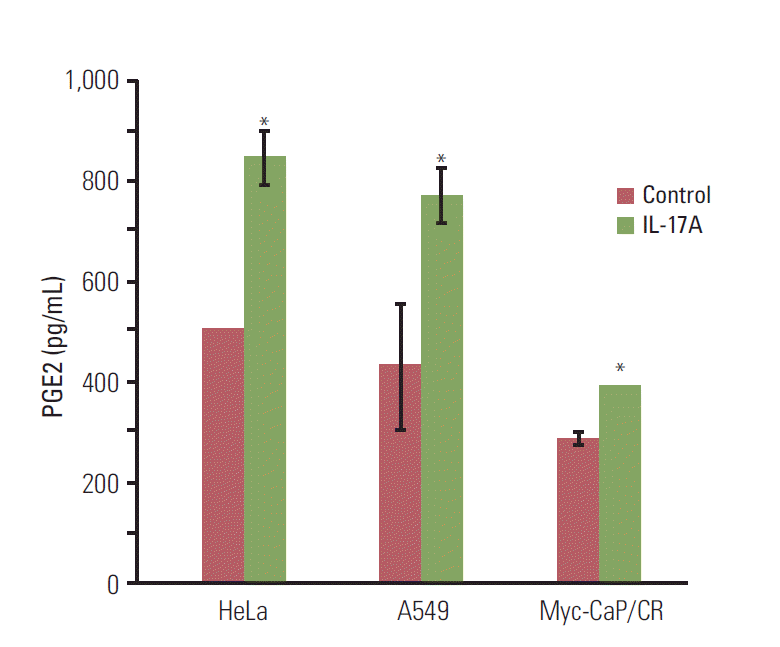 | Fig. 4.Interleukin (IL)-17 induces prostaglandin E2 (PGE2) secretion in cancer cells. The cancer cells were serum starved for 15 hours and then treated without or with 20 ng/mL IL-17A for 24 hours; PGE2 levels were determined using an enzyme-linked immunosorbent assay kit. Values are presented as the mean±standard deviation obtained from three independent experiments. *p < 0.05, compared to the control group.
|
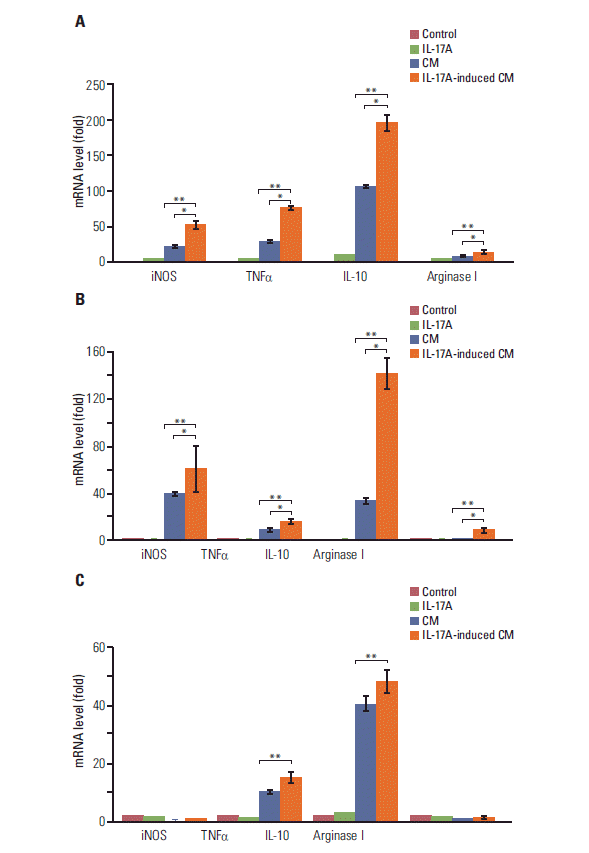 | Fig. 5.Interleukin (IL)-17A-induced conditional medium increases expression of marker genes of M2 macrophages. The conditional medium (CM) or IL-17A-induced CM from human HeLa cancer cells (A) or human A549 cancer cells (B) was used to treat mouse RAW264.7 macrophages; the CM or IL-17A-induced CM from mouse Myc-CaP/CR cancer cells (C) was used to treat human THP-1 monocytes. The control group was treated with serum-free medium that had not been exposed to any cells. The IL-17A group was treated with 20 ng/mL IL-17A in serum-free medium. After 3 hours treatment, mRNA levels of the genes were determined by real-time quantitative reverse transcriptase polymerase chain reaction. Inducible nitric oxide synthase (iNOS) and tumor necrosis factor α (TNFα) are markers for M1 macrophages, and IL-10 and arginase I are markers for M2 macrophages. Values are presented as the mean±standard deviation obtained from three independent experiments. *p < 0.05 or **p < 0.01, compared to the groups as indicated.
|
Go to :
Discussion
Our previous study has demonstrated that IL-17 and PGE2 cooperatively contribute to creation of an M2-macrophagedominant tumor microenvironment in lung cancer [6]. In the present study, our data showed that IL-17 up-regulated COX-2 expression through activation of NF-κB and/or ERK1/2 signaling pathways in three cancer cell lines, namely, HeLa, A549, and Myc-CaP/CR. Subsequently, the increased COX-2 expression stimulated the synthesis and secretion of PGE2 by these cancer cells. Our findings demonstrated that the CM from these cancer cells contained PGE2. The CM induced expression of higher levels of IL-10 than iNOS or TNFα in the monocytes/macrophages, which was more obvious with the IL-17A-induced CM due to the higher levels of PGE2. It is worth pointing out that IL-17A may induce expression of other proteins besides PGE2, which may potentially affect macrophage differentiation. However, our previous study has demonstrated that PGE2 is the key heatstable factor in inducing M2 macrophage differentiation [6]. Because IL-10 is a marker of M2 macrophages [1,2], it is likely that PGE2 in the CM induced M2 macrophage differentiation as we observed previously [6]. Yet, IL-17 did not show any direct effects on the monocytes/macrophages. These findings suggest that IL-17 may indirectly affect M2 macrophage differentiation through induction of PGE2 synthesis in the cancer cells. IL-17 levels are increased in many human cancers such as lung cancer [6] and prostate cancer [13]. Recently, it was reported that TH-17 cell number and IL-17 levels are increased in cervical cancers compared to normal cervical tissues [14]. It has previously been shown that IL-17 promotes growth of human xenograft cervical tumors through recruitment of macrophages to the tumor site [15]. We have previously demonstrated that IL-17, as a chemoattractant, can directly recruit monocytes/macrophages into the tumor microenvironment [6]. In this study, we demonstrated that IL-17 can indirectly induce M2 macrophage differentiation through stimulating cancer cells to secrete PGE2. Therefore, IL-17 appears to play an important role in creation of an M2-macrophage-dominant tumor microenvironment.
IL-17 has been shown to modulate tumor immune microenvironment by recruiting myeloid-derived suppressor cells (MDSCs) and inhibiting CD8+ T cell infiltration at the tumor sites, thus promoting tumor growth [16]. Both MDSCs and M2 macrophages suppress the activity of CD8+ T cells. Therefore, our study is consistent with this previous study in showing IL-17’s role in creating an immunosuppressive tumor microenvironment. Recently, it was reported that IL-17 induces expression of granulocyte colony-stimulating factor that recruits MDSCs to the tumor microenvironment, thereby promoting tumor-resistance to anti-angiogenic therapy [17]. Another study found that blockade of IL-17 increases the cytotoxic activity of CD8+ T cells, and in contrast, eliminates MDSCs and regulatory T cells at tumor sites [18]. IL-17 has been shown to up-regulate expression of prosurvial genes, namely, myeloid cell leukemia sequence 1 (MCL1) and B-cell lymphoma 2 (Bcl-2)-related protein A1 (BCL2A1) in human dendritic cells, thus conferring chemoresistance to 11 chemotherapy agents [19]. High serum levels of IL-17 predict poor prognosis in non-small-cell lung cancer, hepatocellular carcinoma, gastric cancer, breast cancer, and colorectal cancer [20-24]. These recent findings highlight the important significance of IL-17’s function in modulating tumor immune microenvironment.
Go to :
Conclusion
IL-17 up-regulates COX-2 expression in HeLa, A549, and Myc-CaP/CR cancer cells and subsequently increases synthesis and secretion of PGE2 that induces M2 macrophage differentiation. Therefore, IL-17 indirectly promotes M2 macrophage differentiation through stimulation of the COX-2/PGE2 pathways in cancer cells, thus IL-17 plays an indirect role in regulating the tumor immune microenvironment.
Go to :
 XML Download
XML Download