

 PDF
PDF Citation
Citation Print
Print


Introduction
Paclitaxel is a useful drug against a wide range of solid cancers. However, there remains to be a problem of hypersensitivity [1]. Recently, efforts have been focused on developing an oral formulation of paclitaxel. The potential advantages of oral formulation include the following: lack of hypersensitivity, convenience for patients, and potential continuous exposure to paclitaxel. Some oral forms of paclitaxel have been developed and exhibited promising results in phase 1 studies [2,3]; however, none of these agents have been fully developed. The major obstacle in developing oral paclitaxel has been poor oral bioavailability that originates from high affinity for P-glycoprotein (P-gp), which is abundant in the gastrointestinal mucosa [4]. Previous studies illustrated that the oral co-administration of P-gp inhibitors and paclitaxel could increase the bioavailability of paclitaxel [5,6].
Hanmi Pharmaceutical (Seoul, Korea) developed HM30181A, which is a novel inhibitor of P-gp. HM30181A displayed selective inhibition of P-gp and co-administration of oral paclitaxel with it increased the oral bioavailability of paclitaxel in rats [7]. The dose ratio of HM30181A and oral paclitaxel was determined to be 1:2 via the oral absorption test in rats. The tolerability and pharmacokinetics of HM30181A in humans had been evaluated in healthy Korean male volunteers [8]. HM30181A appeared to be well tolerated and had relatively low systemic exposure.
We performed a first-in-human phase I study of an oral drug composed of paclitaxel and HM30181A on patients with advanced solid cancers. The primary objectives of this study are to determine the maximum tolerated dose (MTD), as well as to explore the dose limiting toxicity (DLT). In addition, the secondary objectives are to evaluate the pharmacokinetics and estimate the recommended phase II dose of the study drug in patients with advanced solid cancers.
Materials and Methods
1. Patient eligibility
Patients were considered eligible if they had histologically confirmed advanced solid tumors. The inclusion criteria were as follows: 1) patients for whom no standard therapy exists or who were not amenable to established treatment; 2) between 19 and 70 years of age; 3) Eastern Cooperative Oncology Group performance status of 2 or less; 4) expected life expectancy of at least 12 weeks; 5) adequate hematological, renal, and hepatic function; and 6) no prior chemotherapy, radiation therapy, surgery, and immunotherapy in the previous 4 weeks. Patients with the following conditions were excluded: 1) uncontrolled infectious disease and metastasis to the central nervous system; 2) previous bone marrow transplant; 3) symptomatic atrial or ventricular arrhythmia or congestive heart failure or medical treatment for myocardial infarction within the past 6 months; 4) psychiatric disorders or neurologic problems, including dementia or epilepsy; 5) pregnancy or lactating or with childbearing potential without the use of adequate contraception; and 6) the use of P-gp inhibitors, such as cyclosporine, within 2 weeks prior to study enrollment.
2. Study design
This phase I, open-label, dose-escalating study was conducted at Seoul National University Hospital (Seoul, Korea). This study was performed in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (IRB H-0605-025-174). This study was also registered at http://www.clinicaltrial.gov (NCT01491204).
Paclitaxel solution was started with a dose of 60 mg/m2 and was increased up to 480 mg/m2, in increments of 60 mg/m2. A total of 300 mg of paclitaxel, without Cremophor EL (CrEL) and ethanol, is contained in a 10 mL paclitaxel solution. The dose of HM30181A was half that of paclitaxel. HM30181A was administered with 200 mL water, under an overnight fasting condition, 1 hour before paclitaxel treatment. Each cycle was 28 days, and the test drugs were administered on days 1, 8, and 15 within each cycle. No premedication for hypersensitivity was delivered. Toxicity was graded in accordance to the Common Toxicity Criteria for Adverse Events ver. 3.0 [9]. DLT was defined as follows: 1) grade 4 neutropenia that lasted 7 days in duration or with fever (≥ 38.5°C); 2) grade 3/4 thrombocytopenia for ≥ 7 days in duration or with transfusion or bleeding; 3) treatment delay of > 2 weeks due to drug-related toxicities; or 4) grade 3/4 nonhematologic toxicity (excluding nausea, vomiting, alopecia, anorexia, and fever) that occurred during the first cycle of treatment. In case of toxicity corresponding to DLT after the second cycle, the dose of the following cycle was reduced by 25% of the initial dose. If the hematologic and nonhematologic toxicities were not recovered appropriately by the D1 of the following cycle, or it was necessary to delay dosing for patients’ safety, as determined by the principle investigator, administration could be delayed up to 2 weeks. Patients with absolute neutrophil count (ANC) ≥ 1,500/mm3 and platelet count (PLT) ≥ 100,000 /mm3 were permitted for day 1 dosing, and ANC ≥ 1,000/mm3 and PLT ≥ 75,000/mm3 for D8 and D15 dosing. Dose escalation followed the standard 3+3 rule. The MTD was defined as the dose level; at which, two or more patients experienced DLTs.
Tumor responses were evaluated every two cycles according to Response Evaluation Criteria In Solid Tumors (RECIST) ver. 1.0 [10]. Patients were withdrawn from the study when they displayed signs of progressive disease or unacceptable toxicity, or simply when they withdrew consent.
3. Pharmacokinetics
At least two patients at each dose level underwent blood sampling during the first cycle. The blood sampling times for paclitaxel were immediately before administration, 0.25, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 24, 34, and 48 hours after administration; the blood sampling times for HM30181 were immediately before administration, 0.5, 1, 2, 3, 4, 5, 7, 11, 25, 35, and 49 hours after administration. The plasma concentrations of paclitaxel and HM30181A were measured using a validated liquid chromatography/tandem mass spectrometry method [11]. The actual sample collection time was used for pharmacokinetic analyses. The maximum plasma concentration (Cmax) and time required to reach the Cmax (Tmax) parameter estimates were the actual observed values. The area under the curve (AUC) was calculated by applying a linear-up/logdown trapezoidal method to the observed plasma concentration-time graph. The pharmacokinetic parameters of the test drugs were calculated by using noncompartmental analysis and WinNonlin ver. 5.2 (Pharsight, CA). To evaluate the systemic exposure of paclitaxel according to the dosegroup, we used a noncompartmental analysis because it required little assumption, while providing an exact estimate of pharmacokinetic exposure to individuals. The elimination constant (λz) was determined by a log-linear regression analysis of the terminal phase, and the elimination half-life (T1/2) was obtained by ln(2)/λz. The AUC from time 0 to infinite time (AUCinf) was obtained by summing AUClast (AUC from 0 to the last time to measure the plasma concentration) and AUCext (estimated by taking the ratio between the last measurable plasma concentration and λz). Oral clearance (CL/F) was calculated by dividing the total paclitaxel dose by AUCinf. The volume of distribution (Vd/F) of oral paclitaxel was determined by measuring the ratio of CL/F to λz. The mean residence time was calculated as 1/λz. To evaluate the dose proportionality, the power model (Y=Exp(α) (Dose)β Exp(ε), If beta=1, dose-proportionality can be declared) was applied to each dose-dependent PK parameter (Cmax, AUClast) [12]. All statistical analys es were performed using SAS ver. 9.1 (SAS Institute Inc., Cary, NC).
Results
1. Patient characteristics and treatment
A total of twenty-five patients were enrolled in the study. However, one male patient withdrew his consent without any treatment due to an aggravation from an underlying disease. The baseline characteristics of the remaining 24 patients are shown in Table 1. Three patients with history of adenoid cystic carcinoma underwent radiotherapy-only treatment. The remaining 21 patients were treated with systemic chemotherapies.
Distribution of patients across the cohort and dose escalation scheme are described in Table 2. Three patients were selected to receive each dose level, excluding the 240 mg/m2 dose level, which induced one case of DLT. All patients adhered to the prescribed medications, and the compliance rate was 100%. A total of 83 cycles of paclitaxel were administrated to 24 patients. The median cycle number for paclitaxel was 2.0 (range, 1 to 16) across the entire study population, and the highest cycle number was observed for the 180 mg/m2 dose level (7 cycles; range, 4 to 8).
2. Toxicity
Regarding the paclitaxel dose level of 240 mg/m2, grade 3 neutropenia without fever occurred in a patient with metastatic breast cancer, who was previously treated with four lines of systemic chemotherapies, including paclitaxel. The first cycle could not be completed within 6 weeks in this patient. No other DLT was observed throughout the remaining dose levels; thus, it was not possible to determine MTD.
There were 15 cases of hematologic toxicities and 55 cases of nonhematologic toxicities related to the treatment. Table 3 summarizes the drug-related hematologic toxicities for each dose level. All of the grade 3/4 hematologic toxicities were observed at dose levels of 240 mg/m2 or higher. The most frequent drug-related nonhematologic toxicities included diarrhea (16 patients), nausea (8 patients), and alopecia (7 patients). Table 4 summarizes the drug-related nonhematologic toxicities observed during the study. There was no grade 3 or higher nonhematologic toxicity.
One patient with refractory non-small cell lung cancer, who exhibited multiple lung and bone metastases, experienced a serious adverse event (SAE) at a paclitaxel dose of 420 mg/m2. SAE was cholangiohepatitis, which was recovered after endoscopic biliary drainage. This event had no causal relationship with the investigational product.
3. Pharmacokinetics
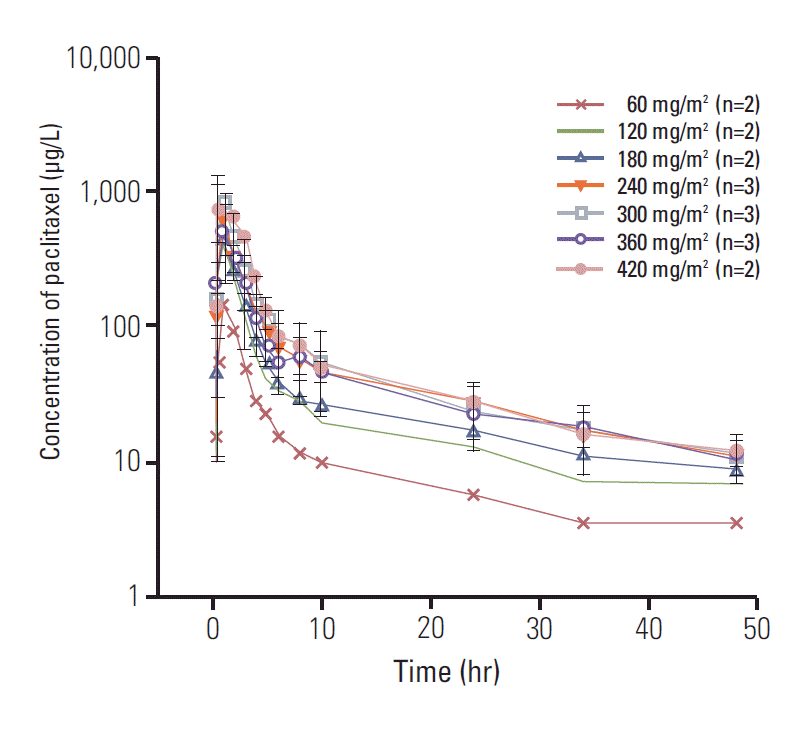
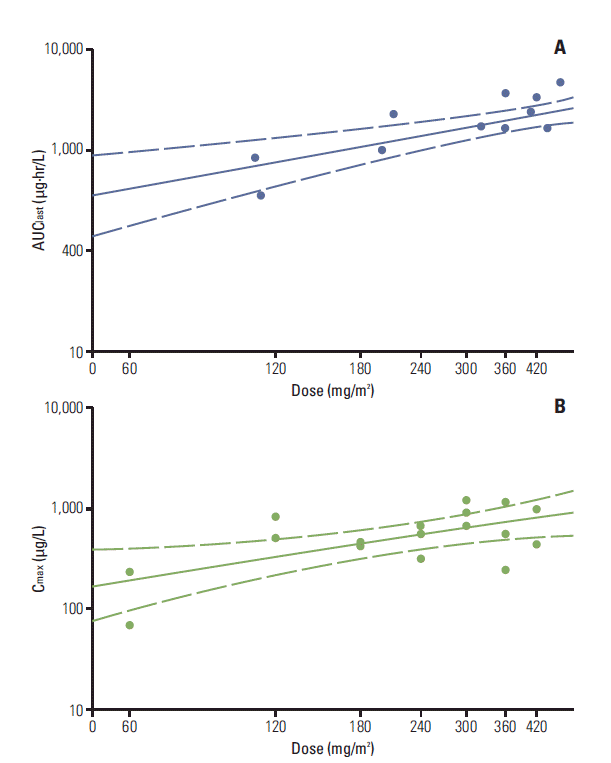
Except the 2 patients who did not consent to collect PK samples and 5 patients whose blood samples were not adequate for analysis, pharmacokinetic evaluations were performed in 17 patients following the first dose over the 60-420 mg/m2 dose range of paclitaxel. The pharmacokinetic parameters of paclitaxel are described in Table 5. Fig. 1 depicts the concentration versus time curves of paclitaxel at each dose level. Paclitaxel was rapidly absorbed, and the peak plasma concentration was achieved within 0.5-1 hour of administration. The T1/2 was 19.9-32.1 hours across all the dose levels (Table 5), which did not show any trend with the dose levels. The mean AUClast values for the doses of 60, 120, 180, 240, 300, 360, and 420 mg/m2 were also shown in Table 5. The maximal Cmax value and the maximal AUClast value were obtained at a dose level of 300 mg/m2 and 420 mg/m2, respectively. Although the dose proportional tendency in pharmacokinetic property was shown using a power model (95% confidence interval of the slope, 0.414 to 1.305 for Cmax and 0.204 to 1.302 for AUClast) (Fig. 2), Cmax and AUClast of paclitaxel did not exhibit a significant increment in response to paclitaxel dose escalation beyond the dose level of maximal Cmax and AUClast. The plasma concentration of paclitaxel over time revealed that the effective range (0.01-0.1 μM) [13] was achieved with doses of 120 mg/m2 or higher for 24 hours.
4. Antitumor activity
Twenty-three patients had measurable diseases. Eight (34.8%) of these patients exhibited stable disease (SD), and 15 patients (65.2%) exhibited progressive disease. Tumor shrinkage was not reported in this study. With regard to each dose level, one, one, three, two, and one patient exhibited SD at 60, 120, 180, 240, and 420 mg/m2. The median duration of SD was 16.5 weeks (range, 12.5 to 62.3 weeks). Among the eight patients who displayed SD, five had adenoid cystic carcinoma, and the remaining 3 had non-small cell lung cancer, rectal cancer, and gallbladder cancer.
Discussion
This is the first-in-human phase I study to determine MTD and explore the safety and pharmacokinetic properties of oral paclitaxel and HM30181A. Although MTD was not determined, this study demonstrated that oral paclitaxel with HM30181A was well tolerated, and therapeutic plasma levels of paclitaxel were achieved.
In this study, Cmax and AUClast exhibited dose linearity in limited range of the dose levels with a power model. However, systemic exposure of paclitaxel was not increased significantly at doses exceeding 300 mg/m2. Considering that T1/2 was not affected across all dose levels, this result could be explained by limited absorption, rather than increased elimination at the dose levels of 360 and 420 mg/m2. Therefore, we judged that MTD would not be reached with additional dose escalation. Additionally, interindividual differences in paclitaxel exposure were prominent in this study. This finding is believed to be due to individual variability of P-gp, which was responsible for the absorption of paclitaxel [14]. When P-gp with high activity is inhibited, paclitaxel absorption will be largely increased. Meanwhile, the inhibition of P-gp with low activity scarcely changes the absorption. Therefore, P-gp variability of each patient could explain the interindividual differences in pharmacokinetics.
The effectiveness of paclitaxel is related to the maintenance of cytotoxic plasma concentrations [15], and the effective cytotoxic plasma concentration range of paclitaxel is 0.01-0.1 μM [13]. In this study, co-administration of oral paclitaxel and HM30181A achieved a therapeutic efficacy at the paclitaxel dose of 120 mg/m2 without significant toxicity. Several studies reported that weekly paclitaxel is highly active and better tolerated than a three-weekly schedule, and the efficacy of weekly paclitaxel was proven even in patients who previously failed with taxanes [16-18]. The superiority of weekly paclitaxel may be due to prolonged drug exposure, direct antiangiogenic effect, or both [19,20]. In the current study, this schedule could be one of the alternatives of weekly paclitaxel in a point of prolonged drug exposure.
The common toxicities of conventional paclitaxel include hypersensitivity, neutropenia, and neuropathy [1]. CrEL is a well-documented risk factor of hypersensitivity reactions, and CrEL-free formulations do not cause hypersensitivity reactions [3,21,22]. Oral paclitaxel and HM30181A did not induce hypersensitivity reactions without premedication, similar to the other CrEL-free paclitaxel formulations. The major determinant of neutropenia could be the length of time in which the upper limit of the paclitaxel therapeutic level (0.1 μM) is exceeded [1]. In this study, the duration of time for which the upper limit of the paclitaxel therapeutic level was exceeded was much shorter than that for which the lower limit of the therapeutic level (0.01 μM) was exceeded. Such property may have contributed to the low incidence of neutropenia in our study. There was only one case of neuropathy in this study: at a dose of 240 mg/m2. CrEL is known to cause neuropathy [23]; however, CrEL-free paclitaxel formulations have also been reported to cause neuropathy at higher levels of paclitaxel exposure [21,22]. These findings indicated that neuropathy could be more likely mediated by paclitaxel itself rather than CrEL. Similar to our findings, another oral formulation of paclitaxel that achieved relatively low plasma levels did not produce neuropathy of grade 3 or higher [3].
The co-administration of oral paclitaxel and HM30181A has some limitations. HM30181A, a P-gp inhibitor, can simultaneously modulate multiple cytochrome P450 enzymes [24]. Hence, HM30181A might alter the pharmacokinetics of other concomitant drugs in an unexpected manner, and the action of HM30181A might be influenced by an interindividual P-gp variation. This possibility must be validated in future studies.
 XML Download
XML Download