

 PDF
PDF Citation
Citation Print
Print


Introduction
Paragangliomas (PGLs), which were first described in 1886 [1], are chromaffin cell tumors that develop from sympathetic and parasympathetic ganglia throughout the abdomen, head and neck area [2]. Some PGLs are inherited, and occur as sporadic tumors. Some hereditary syndromes can be linked to development of PGLs. These syndromes may be von Hippel-Lindau syndrome (VHL) and multiple endocrines neoplasia type 2, and are associated with impaired NF1, RET proto-oncogene or VHL gene expression [3].
In PGL diagnosis, measurement of plasma and urinary metanephrine levels has been used as an effective method. Untreated PGL could have devastating results due to myocardial infarction, severe hypertension, stroke and/or arrhythmia caused by catecholamine excess [2].
Although PGL has been considered as a ten percentage disease (10% metastatic, 10% familial, 10% recurring, 10% extra-adrenal, 10% occurring in children), up-to-date diagnostic techniques illustrate that this rule of 10% does not indeed accurately describe the disease. PGL patients, with 0-36%, may develop metastatic disease. The familial percentage has been revised to approximately 30% [4]. Extra-adrenal PGLs are reported in patients with a range of 15-20% [2]. In general, PGLs are curable, yet, once a patient presents with metastases, the treatment options become limited.
Go to : 
Case Report
In this report, we present the case of a 43-year-old woman with a history of episodic headaches, diaphoresis, and weakness. Hypertensive and tachycardic episodes were also diagnosed. Detailed family history revealed no unexplained or incompletely explained sudden death. She had elevated plasma and urinary metanephrine/catecholamine levels (Table 1), and a right paraaortic mass (9×9.5 cm) was observed on computed tomography (CT). Preoperatively, α- and β-adrenergic receptor blockers were administered. Following successful resection of the tumor, plasma and 24-hour urinary catecholamine levels were normalized. The mass was excised and diagnosis of PGL was confirmed; synaptophysin and chromogranin were positive (Fig. 1). In postoperative follow-up, the patient's blood pressure and catecholamine levels were normalized.
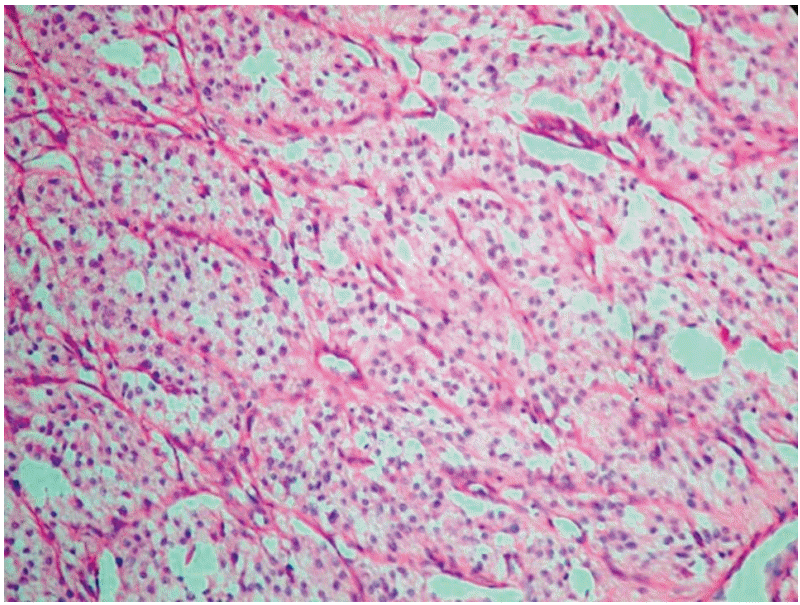 | Fig. 1.Zellballen arrangement of the chief cells with peripherally layered sustentacular cells and a prominent capillary network around each group is noted (H&E staining, ×200).
|
Table 1.
Plasma and urinary catecholamine levels of patients
![]()
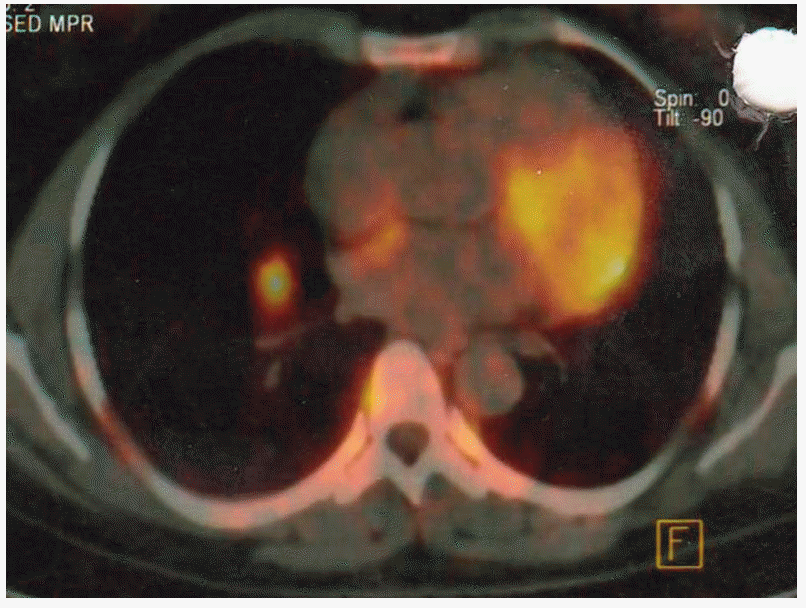
After 20 months, positron emission tomography (PET) showed local recurrence, and metastases were detected in the thorax, abdomen, and skeletal system (Fig. 2). Plasma catecholamine levels were high (Table 1). An anthracycline plus CVD regimen (adriamycine 7 mg/m2, dacarbazine 400 mg/m2, cyclophosphamide 500 mg/m2, vincristine 1.4 mg/m2) on days 1 and 2, and zoledronic acid (4 mg) every 21 days were administered, and no improvement was observed on PET following a treatment of six cycles.
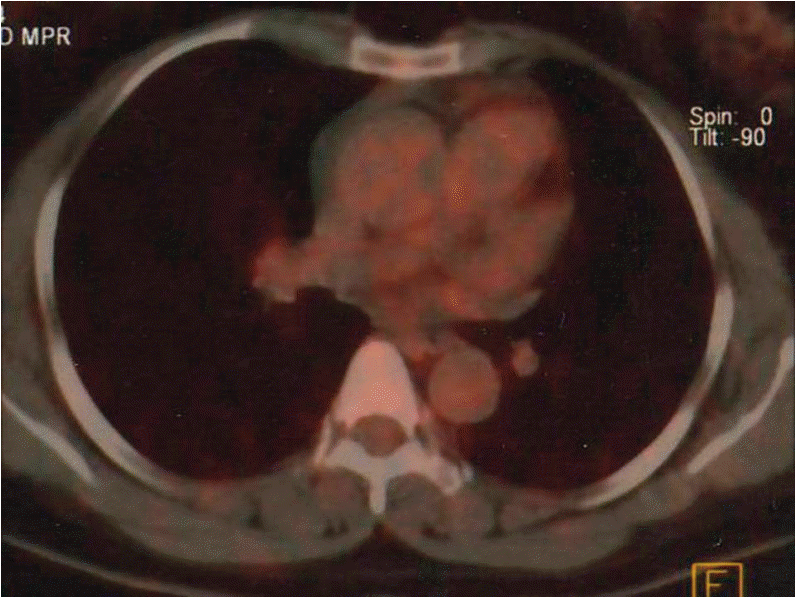
Four months following termination of chemotherapy, calculated plasma and 24-hour urinary catecholamine levels were above normal range. Therefore, sorafenib tosylate (400 mg twice daily) was administered for three months due to recurred-progressive disease. The patient’s plasma and 24-hour urinary catecholamine levels finally decreased, and PET/CT showed that metastatic lesions had regressed after 12 weeks (Table 1, Fig. 3). In addition, the patient’s symptoms decreased. After 64-week treatment with sorafenib, distance and cerebral metastasis occurred, and the patient died.
Go to :
Discussion
PGLs, also called extra-adrenal pheochromocytomas, may develop anywhere from sympathetic/parasympathetic nerve cells to major arteries in the body, with 85-90% of occurrence in the abdomen. These tumors, hypersecreting catecholamines, can be observed in any region from the skull to the pelvis. Although metastatic PGLs (mPGLs) have no reliable markers, they can be characterized as being over 5-cm in mass, invading adjacent structures, and spreading to other organs [2].
Options for treatment of mPGLs include palliative tumor resection, ablation, reduction of tumor burden and excess of catecholamine related symptoms, and cytotoxic hemotherapy or targeted agents [5,6].
The benefits of CVD, the most widely used chemotherapeutic regimen [2], appear to be rather short-termed, and exclude an increase in patients' survival [6]. In a recent case study, another chemotherapeutic agent, temozolomide, was reported to yield positive effects prior to tumor resection [7]. Regarding post tumor removal, treatment with targeted radiotherapeutics may yield significant benefits, the most frequent being treatment with 131I-MIBG. The results here suggested that there was a symptomatic response in as many as 67-98% of patients; an observable tumor shrinkage in 27-47%; and a decrease in secretion in 45-67% [2].
So far, treatment with multiple tyrosine kinase inhibitors (mTKI) like sunitinib, to which five patients showed a partial response and one patient showed clinical improvement, appears to be the most effective approach [8-11]. Sunitinib, an oral multitargeted receptor tyrosine kinase inhibitor with antiangiogenic and antitumor activity, targets platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), KIT, and FLT3. Duration of response on sunitinib was 24, 40, 40, 24, 4, and 24 weeks, respectively, in all patients.
Phosphatidylinositide 3-kinase/Akt/mammalian target of rapamycin (mTOR) pathway in the pathogenesis of malignant neuroendocrine tumors includes pheochromocy-toma. Everolimus is a compound that inhibits mTOR signalling. Treatment with everolimus, as targeted therapy, was attempted, however, no significant benefits were obtained in four patients [12].
Imatinib does not appear to be useful for treatment of mPGLs; progression was reported in two patients who were administered this drug [13].
Sorafenib, a mTKI, is used in treatment of advanced renal cell and hepatocellular carcinomas. It is an inhibitor of RAF kinase, c-KIT, FLT-3, RET, VEGFRs (VEGFR-2, VEGFR-3), and PDGFR (PDGFR-β). It reduces activation of MEK and MAP kinase, and causes inhibition of cell proliferation, invasion, migration, and tumor growth.
In our case, the patient obtained clinical and laboratory benefits from sorafenib by showing a truly complete response after a three-month therapy. Response duration of sorafenib was 64 weeks in this case. Considering other cases in which sunitinib was administered, this period is quite important. Sorafenib may have potential for treatment of mPGLs since it targets several cellular pathways, one of which is VEGFR-2, -3, and possibly others. High vascularization is usually a common characteristic in almost all metastatic diseases [2]. Since this is the case, sorafenib may prove to be beneficial in inhibiting vascularization. In addition, expression of the RET gene mutation is known to be the most frequent type in familial PGLs [3]. Sorafenib may also be an active agent in inhibition of the RET protooncogene for familial PGLs.
To the best of our knowledge, in the literature, there are no cases with mPGLs subjected to treatment utilizing sorafenib. Because the options for treatment of mPGLs are rather limited, sorafenib may prove to be a very effective agent. The result we obtained with our case is very exciting, yet there is still a need for comprehensive research investigating reliability of this drug in treatment of patients with mPGLs.
Go to :
 XML Download
XML Download