

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC) has been a significant cause of morbidity and mortality in the world [1]. Although patients diagnosed with early stage disease have a high cure rate, many present later when five year survival is poor. Treatment of CRC has shown significant improvement in recent years, with new generation chemotherapeutic agents and molecular targeted agents.
During the 1990s, the introduction of oxaliplatin and irinotecan to the therapeutic repertoire for advanced CRC resulted in clear benefits for patients [2,3]. CRC carcinogenesis and biology have recently been recognized as multistep processes involving accumulation of molecular alterations [4]; in addition, it has also been suggested that associations may exist between many of these abnormalities and patient survival [5,6]. Increased understanding of the changes in specific molecular pathways that are responsible for disease progression and poor prognosis may prove essential in development of more effective targeted therapy. The two most relevant targets for biologic agents are the epithelial growth factor receptor (EGFR) and the vascular endothelial growth factor (VEGF). Kirsten-ras (KRAS) is a proto-oncogene encoding a small 21 kD guanosine triphosphate/guanosine diphosphate binding protein involved in regulation of cellular response to many extracellular stimuli [7]. Mutations within KRAS abrogating GTPase activity and resulting in activation of RAS/RAF signaling are found in 35% to 42% of CRCs and are thought to occur early in CRC carcinogenesis. In addition, mutation of KRAS is predictive of nonresponse to EGFR-targeted monoclonal antibody therapy across all treatment lines, either as a single agent or in combination chemotherapy [8,9]. Thus, determination of KRAS status is now recommended in patients with advanced CRC who are selected for EGFR targeted therapies. However, although EGFR targeted therapies have shown clinical benefit in advanced CRC, both with modest but definite activity, but also significant unwanted adverse effects, the cost has still, to some extent, restricted the use of EGFR targeted therapies.
The question of whether KRAS mutation in CRC has a prognostic role, independent of anti-EGFR therapies has been controversial [10,11]. Previous studies have not been conclusive, even among several large studies [12,13]. In addition, whether or not KRAS mutational status affects the outcome of oxaliplatin- or irinotecan-based chemotherapy remains uncertain.
We intended to evaluate the role of the status of KRAS mutation as a prognostic marker in CRC independent of anti-EGFR therapies. In addition, we wanted to determine whether KRAS mutation is a predictive biomarker for oxaliplatin- or irinotecan-based (CPT-11) chemotherapy in CRC.
Materials and Methods
1. Patients
We retrospectively reviewed the records of 103 CRC patients who were available for evaluation of KRAS mutation status and had been treated with systemic chemotherapy at the Korea University Anam Hospital, Seoul, Korea between April 2004 and January 2011. All patients analyzed had pathologically or cytologically proven metastatic or recurrent CRC. During the treatment course, patients had not received any anti-EGFR therapies; however, some patients had been treated with chemotherapy including anti-VEGF agents. The following clinical data were collected from the medical records of each patient: physical examination, surgical and pathologic reports, and imaging. Medical information, including chemotherapy regimens, response, date of progression, last follow-up, and deaths was collected.
2. Chemotherapy
The decision regarding whether or not chemotherapy was conducted depended, in all cases, on discussion between physician and patient. The chemotherapy regimen to be used was determined by the physician. As first-line chemotherapy, oxaliplatin plus intravenous or oral 5-fluorouracil (5-FU) combinations (fluorouracil, leucovorin, and oxaliplatin; [FOLFOX] or capecitabine plus oxaliplain [XELOX]) with or without anti-VEGF agents have usually been proposed to patients. As second-line chemotherapy, irinotecan-single or plus 5-FU (5-fluorouracil, leucovorin, and irinotecan [FOLFIRI]) with or without anti-VEGF agents have usually been used. Chemotherapy was repeated every two weeks, according to protocol. All tumor measurements were assessed after every three or four cycles of chemotherapy, using computed tomography scan and other tests that were used initially in staging of the tumor. Responses were classified according to the Response Evaluation Criteria in Solid Tumors (RECIST) ver. 1.0.
3. Mutation analysis
DNA was extracted from five paraffin sections of 10 µm thickness containing a representative portion of tumor tissue (Qiagen, Hilden, Germany). Fifty nanograms of DNA were amplified in a 20 µL reaction solution containing 10 µL of 2× concentrated HotStarTaq Master Mix (Qiagen), including polymerase chain reaction (PCR) buffer, 3 mM MgCl2, 400 µM each of dNTP, and 0.3 µM each of the primer pairs (codon 12, 13; F: 5'-CGTCTGCAGTCAACTGGAAT, R: 5'-GAGAATGGTCCTGCACCAGTAA). Amplifications were performed using a 15-minute initial denaturation at 95℃, followed by 35 cycles of 30 seconds at 94℃, 30 seconds at 59℃, and 30 seconds at 72℃, and a 10-minute final extension at 72℃. The PCR products were then 2% gel-purified using the QIAgen gel extraction kit (Qiagen).
DNA sequencing was performed as follows: first, digested mutated DNA was used as a template for the second PCR, in which the primer Ras 3 antisense; 5'-GGATGGTCCTCCACCAGTAATATGGATATTA-3') was used instead of Ras 2 (3'). The PCR was run under the same conditions as the first PCR for 32 cycles. Because of the nested antisense primer (Ras 3), the second PCR generated a fragment of 152 bp. This mutated DNA was excised from 3% agarose gels. The amplicons were then purified using the High Pure PCR Product Purification kit (Boehringer-Mannheim, Mannheim, Germany). Five nanograms of the purified amplicons was used for sequencing, which was performed using the Big Dye RR Terminator reaction (ABI, Weiterstadt, Germany). The product was run on a 5% polyacrylamide gel in an ABI 373A Sequencer (ABI) and analyzed for point mutations of the respective amplicons.
4. Statistical analysis
Treatment outcomes were estimated as response rate (RR), progression free survival (PFS), and overall survival (OS). Tumor response was determined according to RECIST ver. 1.0. PFS was defined as the time from the date of the diagnosis for recurrence or metastatic disease to the date of disease progression or death from any cause. OS was defined as the time between the date of the diagnosis for recurrence or metastatic disease and the date of death from any cause. The PFS for oxaliplatin- and irinotecan-based chemotherapy and the OS according to KRAS status were analyzed using the Kaplan-Meier method to estimate the probability of survival and survival difference with the use of the log-rank test. The χ2-test or Fisher's exact test was used for comparison of categorical variables. All reported p-values were the result of two-sided tests, with p<0.05 considered statistically significant.
Cox proportional hazards regression model was employed in univariate and multivariate analyses for identification of the significant independent prognostic factors of various clinical parameters for survival. Significant prognostic variables in univariate analysis for OS were included in multivariate analysis. p-value less than 0.05 was considered statistically significant.
Results
1. Patients' characteristics
Of all patients diagnosed as metastatic or recurrent CRC between April 2004 and January 2011, 103 were available for evaluation of the status of KRAS mutation and had received oxaliplatin based combination as a first-line therapy. Among 92 patients who experienced disease progression after starting first-line chemotherapy, 82 (89.1%) patients received treatment with irinotecan-based combination chemotherapy. None of the patients had received any anti-EGFR therapies during their treatment course. KRAS mutations were detected in 48.5% of tested patients. A summary of the patients' characteristics according to KRAS mutational status is shown in Table 1. The median age of patients was 61 years (range, 20 to 85 years) at diagnosis, and the male/female ratio was 1.3/1.0. Characteristics of patients were generally similar between the KRASmutation and the KRAS wild type groups. Anti VEGF therapy had been used more frequently in the KRAS mutation group, as compared to the KRAS wild type group during the treatment course (p=0.001).
2. Outcomes for chemotherapy
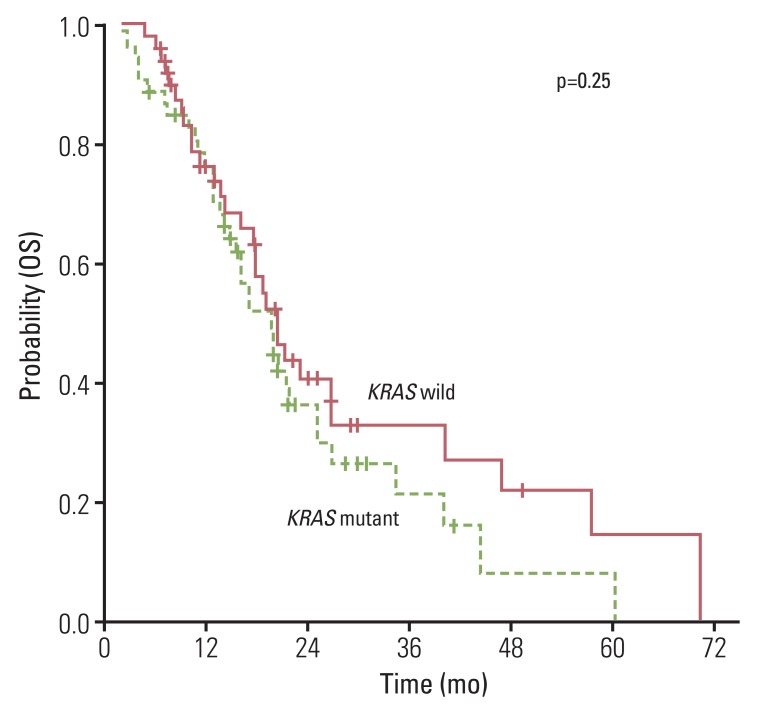
The overall RR of FOLFOX or XELOX was 37.8% and the disease control rate was 76.7% (Table 2). No significantly different response was observed for oxaliplatin (p=0.66) the status of KRAS mutation. Median PFS for first-line chemotherapy was 6.2 (95% confidence interval [CI], 5.22 to 7.18). No significant difference in PFS was observed between the KRAS mutation group and the wild type group (5.7 months; 95% CI, 2.91 to 8.4 months vs. 6.2 months; 95% CI, 5.39 to 7.01 months; p=0.58) (Fig. 1A). In the second-line therapy, the overall RR of CPT-11 and FOLFIRI was 10.9% and the disease control rate was 60.9%. According to the status of KRAS mutation, there was no significant difference of RR and PFS to CPT-11 and FOLFIRI (Table 2, Fig. 1B). In addition, patients with KRAS mutation showed similar OS, as compared to patients with no mutation (Fig. 2).
3. Prognostic analysis
Results of prognostic analysis are shown in Table 3. In assessment of KRAS as a prognostic marker, results of univariate and multivariate analysis showed no evidence of an effect on PFS to oxaliplatin or irinotecan (oxaliplatin: hazard ratio, 0.892; 95% CI, 0.590 to 1.347; p=0.586 and irinotecan: hazard ratio, 0.831; 95% CI, 0.524 to 1.319; p=0.433). Similarly, there was no evidence indicating that KRAS mutation was a prognostic factor for OS (hazard ratio, 0.754; 95% CI, 0.460 to 1.236; p=0.263). In addition, anti-VEGF therapies did not affect PFS to oxaliplatin or irinotecan and OS.
Discussion
In this study, we posed two interesting questions. The first question, whether or not KRAS mutations are prognostic in metastatic or recurrent CRC independent of anti-EGFR therapies. The second question, whether or not KRAS mutations affect the treatment outcome from irinotecan or oxaliplatin.
The predictive and prognostic value of KRAS mutation in a frontline or refractory treatment setting was confirmed by retrospective analysis of the phase III trial, including anti-EGFR therapy [9,14]. An absolute difference was observed between treatments with and without anti-EGFR therapy in terms of a higher RR and PFS in the KRAS wild type cohort. A benefit in terms of the median OS time was observed in the investigational arm containing anti-EGFR therapy in KRAS wild type tumors. However, the prognostic value of KRAS mutation remains controversial independent of anti-EGFR therapies [15,16]. Our data demonstrated that KRAS tumor mutation status has no major prognostic value for OS in patients with advanced CRC treated with cytotoxic chemotherapy. This finding was in accordance with data from other smaller retrospective studies [17,18]. However, simultaneously, conflicting findings were reported in the two large collaborative Kristen Ras in Colorectal Cancer Collaborative Group (RASCAL) studies of 2,721 and 4,268 patients with CRC, respectively [19,20]. While the first RASCAL study reported an increased risk of recurrence and death linked to KRAS mutation, the second study refined this observation to report significant prognostic value in failure free survival only with the G12V mutation in Dukes' C patients. Thus, because many other factors might affect our finding, the clinical impact of our results cannot be concluded. This study was also a retrospective analysis with a small sample size.
In addition, the prognosis of CRC was affected by mutational status of many other genes as well as KRAS. Tumorigenesis and tumor progression of CRC result from multiple genetic and epigenetic abnormalities, including defective DNA mismatch repair and mutation of KRAS, NRAS, BRAF, PI3K, PIK3CA, and p53 [21-23]. These various genetic and epigenetic changes may affect the survival of patients with CRC; however, in this study, we focused only on the status of KRAS mutation for analysis of the survival of CRC.
For CRC patients with KRAS mutation, it is also important to determine whether these mutations, as well as predicting benefit from anti-EGFR therapies, may affect the ability to benefit from other cytotoxic agents or the prognosis independent of treatment. This information is indispensable to development of guidelines for individual medicines in order to avoid overtreatment and undertreatment. In this study, we found that RR and PFS for oxaliplatin or irinotecan did not differ according to the status of KRAS mutation. This finding was similar to that of a previous study. Richman et al. [24] also reported that KRAS/BRAF was not a predictive biomarker for irinotecan or oxaliplatin. However, that study revealed an association of KRAS/BRAF mutation with poor prognosis. This discrepancy might be derived from the heterogeneity of tumor and patients between Richman's [24] and our study. In addition, we found that there was no difference in RR between oxaliplatin- and irinotecan-based chemotherapy in patients with KRAS mutation (p=0.67).
Personalized medicine was applied to patients with CRC. On the basis of data outlined in the introduction, drug licenses have been amended, and many patients are now being offered KRAS testing with a view to receiving anti-EGFR therapies if their tumor is KRAS wild type. However, due to the high cost of anti-EGFR agents, many patients still receive only cytotoxic chemotherapy without molecular targeted agents. Thus, it is important to have established patients' response to standard cytotoxic chemotherapies according to the status of KRAS mutation. Findings of this study demonstrated that KRAS mutation is not a prognostic marker for PFS to oxaliplatin or irinotecan and OS in CRC patients who did not receive anti-EGFR therapies. In other words, although CRC patients with KRAS mutation cannot benefit from anti-EGFR therapies, they may still benefit from standard cytotoxic chemotherapies.
The potential for personalized therapy is not confined to anti-EGFR therapies. In order to realize personalized medicine in CRC patients, the oncologist must evaluate the relation between various individual candidate markers and gene signatures and the effect of fluoropyrimidines, oxaliplatin, irinotecan, bavacizumab, and many new novel agents. The first step in this research is bio-banking of tumor and blood samples from patients.
Conclusion
Mutation of KRAS is predictive of nonresponse to EGFR-targeted monoclonal antibody therapy across all treatment lines, either as a single agent or in combination chemotherapy. However, the question of whether KRAS mutation in CRC has a prognostic role, independent of anti-EGFR therapies has been controversial. Furthermore, whether or not KRAS mutational status affects the outcome of oxaliplatinor irinotecan-based chemotherapy remains uncertain. Based on this study, the response for oxaliplatin- and irinotecan-based cytotoxic chemotherapy did not differ according to the KRAS mutation status. In addition, KRAS mutation is not a prognostic marker for PFS to oxaliplatin or irinotecan and OS in CRC patients who did not receive anti-EGFR therapies.

 XML Download
XML Download