

 PDF
PDF Citation
Citation Print
Print


Introduction
Angiomatoid fibrous histiocytoma (AFH) is a rare soft-tissue tumor, typically occurring in the extremities of children and young adults [1,2]. It presents as a painless, slow growing subcutaneous mass, and, on clinical examination, it is thought to be representative of lymphadenopathy, or a benign cyst. Owing to its local aggressiveness but low propensity to metastasize, it has been classified by the World Health Organization as a fibrohistiocytic tumor of intermediate malignancy [2]. The histology of this tumor is characterized by a fibrous capsule, surrounding lymphocytic infiltrate, and blood-filled cystic spaces lined with flattened tumor cells [1]. Although little is known about the molecular mechanisms involved in development of AFH, three genetically investigated cases of AFH have recently been reported [3-5]. Only two previous cases of AFH as a second tumor have been reported [6,7]. This is a case report on AFH occurring as a second tumor in a young adult who underwent treatment for testicular cancer.
Go to : 
Case Report
A 27-year-old man was admitted with a palpable abdominal mass in May 2009. Chest and abdominopelvic computed tomography (CT) scan showed enlarged para-aortic lymph nodes measuring 9 cm in size, including supraclavicular, mediastinal, and abdominal lymph nodes. Multiple liver and lung metastases were also observed. Levels of serum beta-human chorionic gonadotrophin (beta-hCG), alpha-fetoprotein (AFP), and serum lactate dehydrogenase (LDH) were elevated (beta-hCG, 175,910 mIU/mL; AFP, 109.5 ng/mL; LDH, 475 IU/L). A positron emission tomography (PET)-CT scan showed multiple areas of strong fluorodeoxyglucose (FDG) uptake in the aforementioned lesions. Percutaneous lung biopsy for metastasis confirmed the histological diagnosis of seminoma. Ultrasonography of the testis showed a diffuse infiltrative lesion of the right testis. The patient underwent right orchiectomy. Pathologic examination also revealed a seminoma. Although the diagnosis of seminoma was confirmed in lung and orchiectomy specimens, because of the elevated level of AFP, it was considered to be mixed germ cell tumor (GCT) with seminoma. Based on chest CT, abdominopelvic CT, PET scan, and tumor markers, the patient's state corresponded to stage IIIC and the high risk group according to the American Joint Committee of Cancer (AJCC) staging system and International Germ Cell Cancer Collaborative Group (IGCCCG) classification. After undergoing right orchiectomy, he received primary chemotherapy with bleomycin, etoposide, and cisplatin (BEP). After four cycles of BEP (July 2009), on the subsequent chest and abdominopelvic CT scan, metastatic nodules in the lung and liver had almost disappeared. However, the mediastinal and para-aortic lymph nodes, which had obviously decreased in size, were still evident. The largest residual mass was a para-aortic lymph node, which was seen as a thin-walled, cystic mass, measuring 3.2 cm×2.3 cm. A follow-up PET-CT scan showed no FDG uptake in the residual masses. Levels of beta-hCG, AFP, and LDH were normalized. According to the guidelines, surgical resection should be performed at all sites of residual mass. However, due to the presence of multiple lesions, which were scattered throughout the thorax and abdomen, removal of all residual masses was impossible. Therefore, the patient received two additional cycles of etoposide and cisplatin (EP) and the treatment was completed. Chest CT, abdomen CT, and PET-CT showed no significant change in the residual masses and no newly developed lesions were observed during the following year. At 16 months after chemotherapy (December 2010), a mass in the left supraclavicular area was observed on follow-up neck CT. The mass was a well-defined enhancing lesion with an internal necrotic or cystic component measuring 3×2.2×2 cm in size (Fig. 1A). This lesion did not show FDG uptake on PET-CT (Fig. 1B). The other residual masses had decreased in size. The levels of all tumor markers were consistently within normal limits (beta-hCG, <0.1 mIU/mL; AFP, 3.3 ng/mL; LDH, 171 IU/L). The supraclavicular mass, which had been growing for the last three months, was suspected of being metastatic and was surgically resected. Histological examination revealed a subcutaneous, fibrohistiocytic proliferation with cystic spaces and a pseudocapsule, surrounded by lymphoid tissue (Fig. 2A). At high power, the tumor cells were spindle or epithelioid in shape, and were arranged in a nodular pattern (Fig. 2B). In immunohistochemical staining, the tumor cells were positive for CD68 (Fig. 2C) and desmin (focal) (Fig. 2D), and negative for epithelial membrane antigen, S-100, CD34, and human melanoma black 45. Based on the morphology and results of immunohistochemical staining, a diagnosis of AFH was made. He did not receive subsequent chemotherapy or radiation therapy. Relapse of the tumor did not occur within 12 months.
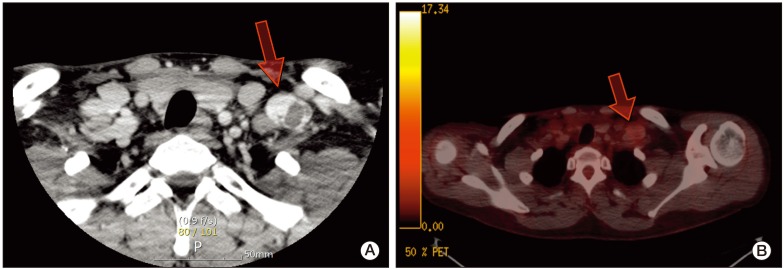 | Fig. 1(A) Enhanced neck computed tomography showed a well-defined enhancing mass with an internal cystic component in the left supraclavicular area (arrow). (B) Positron emission tomography-computed tomography showed no definite fluorodeoxyglucose uptake in the lesion (arrow).
|
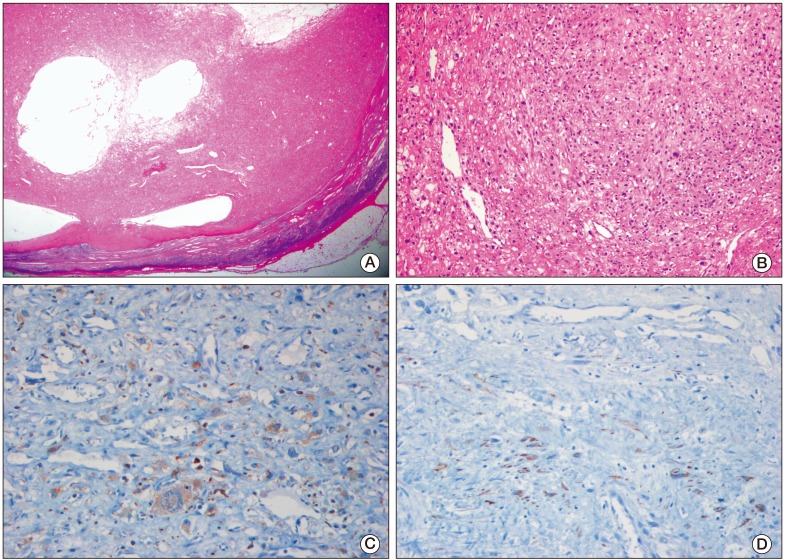 | Fig. 2(A) Low magnification microscopic photograph of the tumor demonstrated fibrohistiocytic proliferation with cystic spaces and a pseudocapsule, surrounded by lymphoid tissue (H&E staining, ×10). (B) The tumor cells were spindle or epithelioid in shape, and were arranged in a nodular pattern (H&E staining, ×100). (C) The tumor cells were positive for CD68 (×200). (D) The tumor cells were focally positive for desmin (×200).
|
Go to :
Discussion
AFH, which was first described by Enzinger [1] in 1979, has several histopathologic findings, such as 1) solid arrays or nests of histiocyte-like cells, 2) hemorrhagic cyst-like spaces, and 3) aggregates of chronic inflammatory cells. Clinically, AFH is a rare soft tissue tumor with a low-grade malignancy, which occurs primarily in children or young people, and involves the extremities and trunk [1,8]. In a large study on AFH reported by Costa and Weiss [2], development of local recurrence occurred in 12% of patients and only 4% of patients deveoped metastasis. Immunohistochemical studies are helpful in differential diagnosis of AFH, which is characteristically often positive for CD68, CD99, epithelial membrane antigen (EMA), and desmin, and negative for endothelial markers, such as factor VIII-related antigen and CD34 [8]. In this case, the tumor cells were positive for CD68 and desmin, and negative for EMA. However, no immunohistochemical stain of CD99 was performed. Several recent cytogenetic and molecular studies in AFH have reported three specific gene rearrangements, involving the FUS-ATF1, EWSR1-ATF1, or EWSR1-CREB1 fusion genes [3-5]. Although we did not investigate these molecular alterations in our case, the diagnosis of AFH could be made comfortably on the basis of morphology coupled with the immunohistochemical findings, except EMA and CD99. Association between AFH and other diseases has not been reported, except in a child infected with human immunodeficiency virus [9]. In our case, the patient was not immunocompromised because the chemotherapy had been completed at that time. Two cases of AFH as a second tumor have been reported, in an 18-year-old girl with a four-year history of treatment for Hodgkin's disease [6], and in a 10-year-old boy with neuroblastoma [7]. This is the first reported case of AFH as a second neoplasm in a patient with testicular cancer.
Testicular cancer is a relatively uncommon tumor comprising only 2% of all human malignancies, and is a largely curable malignancy, with a five-year relative survival rate of more than 90% [10]. Like AFH, testicular cancer is more common in adolescents and young adults. Most primary testicular tumors originate from germ cells. GCTs are classified as seminoma or nonseminoma. Prognosis of pure seminoma is generally more favorable than that of nonseminoma. In contrast, nonseminoma is the more clinically aggressive tumor. Serum tumor markers, such as AFP, beta-hCG, and LDH, are important for diagnosis and treatment of GCTs. AFP is a serum tumor marker produced by nonseminomatous cells. When its histology is that of a seminoma with an elevated serum AFP, management follows that for a nonseminoma. Our case, which had the histology of a seminoma, because of an elevated level of serum AFP, was treated as a nonseminoma. At the conclusion of initial chemotherapy, patients with a residual tumor need to undergo further evaluation. In seminoma, serum tumor markers and PET scan are helpful in assessment of residual tumor viability [11]. A PET scan has a high positive and negative predictive value for the remaining viable tissues in patients with residual masses after chemotherapy for seminoma. In nonseminoma, unlike in seminoma, PET scans for residual disease have a limited predictive value [12]. Therefore, even though multiple masses are still evident and tumor markers are negative after primary chemotherapy, the National Comprehensive Cancer Network (NCCN) guidelines have recommended surgical resection for all residual disease. In our case, we considered surgical resection for all residual masses. However, not all masses could be resected, because they were scattered extensively throughout the thorax and abdomen. Instead, we treated the patient with additional cycles of chemotherapy and performed regular follow-up with CT, PET scan, and serum tumor markers. Newly developed masses were immediately resected during the follow-up period. Consequentially, our case emphasized the importance of pathologic confirmation of the recurred mass in testicular cancer obtained by surgical excision.
In testicular cancer, which affects young men and has a good prognosis, concerns have been raised regarding the risk of secondary malignancy after exposure to chemotherapeutic agents and irradiation. In a large scale cohort study [13], long-term survivors of testicular cancer had a statistically significant increased risk of second malignancy. In the current case, the role of previous chemotherapy in development of AFH remains unclear. Because AFH and GCT are common in adolescents or young adults, the occurrence of AFH in the setting of GCT could be a mere coincidence. However, it is not a coincidence that development of both AFH and GCT, which are rare tumors, occurred within a short interval.
In treatment of AFH, because it showed nearly benign behavior, complete surgical excision without adjuvant therapy is the treatment of choice [14]. The role of adjuvant treatment is not clear. According to some reports, radiotherapy was effective in cases with positive resection margins or in recurrent disease [15]. We did not administer any additional treatment in this patient, and, since then, he has had no recurrences.
We report on the first case of a AFH occuring as a second tumor in a young adult with testicular cancer. Conduct of additional studies will be needed in order to examine the association between AFH and testicular cancer or secondary malignancy after chemotherapy.
Go to :
 XML Download
XML Download