

 PDF
PDF Citation
Citation Print
Print


Introduction
Diagnostic pathology has shown remarkable improvement in recent years, and routine use of immunohistochemistry and electron microscopy has enabled more precise diagnoses. The availability of improved pathological methods for diagnosis of neuroendocrine tumors has also resulted in the recent recognition that these neoplasms have a wider spectrum. The incidence of unknown primary neuroendocrine tumors has not been determined; however, they have been estimated to account for approximately 3% of all malignancy of unknown primary sites [1]. These tumors, which represent a subset of unknown primary carcinomas, have a relatively favorable prognosis. However, poorly differentiated neuroendocrine carcinoma (PDNEC) has an aggressive biology and the median survival duration is only 10 months [1-3]. Most of these patients had several sites of metastasis, often with predominant tumor in bones, liver, and lymph nodes (particularly the retroperitoneal and mediastinal nodes). However, involvement of a single lymph node (LN) is very rare [1,4]. Here, we present a case of PDNEC in a perigastric LN only with no identifiable primary site.
Go to : 
Case Report
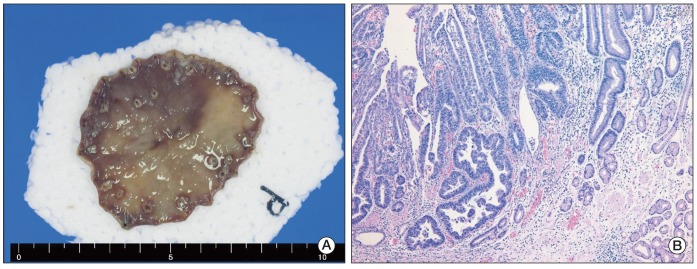
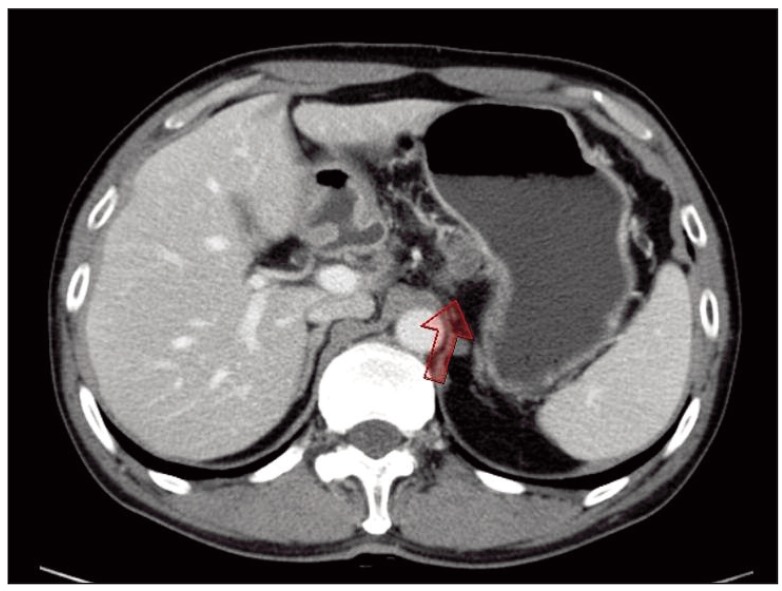
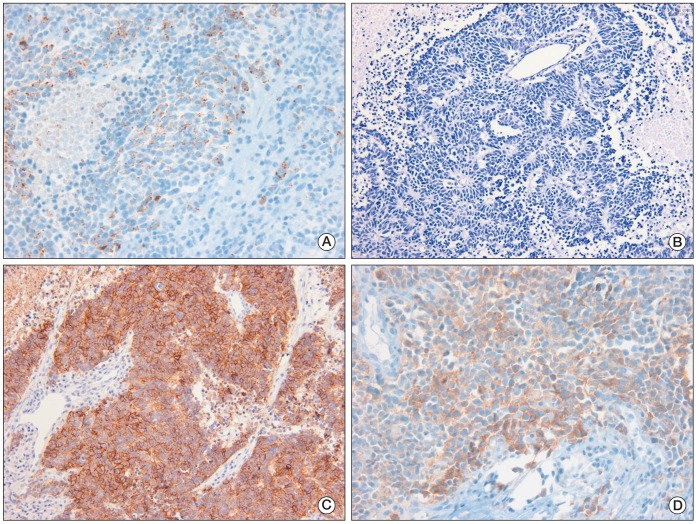
A 52-year-old male patient was diagnosed as having early gastric cancer at the lesser curvature of the antrum by routine esophagogastroduodenoscopy in June 2008. He had no relevant previous medical history. Results of a subsequent endoscopic biopsy resulted in a histological diagnosis of well-differentiated adenocarcinoma. Computed tomography (CT) of the abdomen showed no evidence of LN enlargement or distant metastases. He was diagnosed as having type IIc early gastric cancer, which met the indications for endoscopic submucosal dissection (ESD). ESD was performed successfully (Fig. 1A), and the resected specimen confirmed the presence of well differentiated adenocarcinoma, with negative resection margins, and the absence of lymphovascular or perineural invasion by hematoxylin and eosin (H&E) staining (Fig. 1B). However, six months later, a LN enlargement measuring 2.0 cm at the lesser curvature side of the stomach was observed on a routine abdominal and pelvic CT scan (Fig. 2). He had no symptoms, and physical examination failed to reveal evidence of peripheral lymphadenopathy, and a meticulous skin examination revealed no suspicious lesions. He underwent subtotal gastrectomy and regional LN dissection under a suggestive preoperative diagnosis of gastric adenocarcinoma with perigastric LN metastasis. Microscopically, no residual tumor was found in the stomach, and the pathologic findings of the single perigastric LN differed completely from those of gastric adenocarcinoma. It was presented to PDNEC by morphology on H&E stain (Fig. 3A). Results of immunohistochemical staining showed that tumor cells were positive for cytokeratin 20 (1 : 200, Leica, Newcastle-upon-Tyne, UK), CD56 (1 : 150, Leica) (Fig. 3B), synaptophysin (1 : 200, NeoMarkers, Fremont, CA) (Fig. 3C), and chromogranin (1 : 100, Leica) (Fig. 3D), but negative for cytokeratin 7 (1 : 600, NeoMarkers), thyroid transcription factor-1 (1 : 300, Leica), S-100 (1 : 800, Dako, Glostrup, Demark), and human melanoma black 45 (1 : 150, Leica). Therefore, based on histological and immunohistochemical findings, a diagnosis of PDNEC was made. We performed immunohistochemistry in order to rule out neuroendocrine differentiation in previous well-differentiated adenocarcinoma from the ESD specimen; according to the results, all of the tumor cells were negative for CD56, synaptophysin, and chromogranin. In addition, when the ESD specimen and the specimen resected after subtotal gastrectomy were mapped, no other primary site was identified in the stomach specimen. In our search for the primary site, we also performed chest CT, bronchoscopy, colonoscopy, and a positron emission tomography-CT scan. After an extensive work up, no primary site was identified and no further metastatic lesions were found. He was diagnosed as having PDNEC of unknown origin in one perigastric LN. He also received four cycles of etoposide and cisplatin. He has remained disease-free for more than two years after surgical resection. Written informed consent was obtained from the patient for report of this case.
Go to :
Discussion
This report described a case of PDNEC in a single perigastric LN from a presumed unknown primary site. There are two models of tumor progression for these nodal neuroendocrine carcinomas from an unknown primary site [5,6]: nodal neuroendocrine carcinoma originates from the LN via formation of malignant precursor cells; and it originates from a spontaneously regressed primary lesion. According to a study reported by Kuwabara et al. [7], primary nodal neuroendocrine carcinoma follows a less aggressive course than its metastatic nodal involvement. If nodal neuroendocrine carcinoma is the primary disease, wide surgical resection is the treatment of choice for localized disease. On the other hand, if nodal neuroendocrine carcinoma is formed by systemic metastasis from an unknown primary site, in view of the aggressive behavior of this tumor, platinum-based adjuvant chemotherapy is required after surgical resection. However, determination of whether nodal neuroendocrine carcinoma is primary or metastatic at initial presentation is difficult, thus, localized therapy combined with chemotherapy and regular close follow-up is essential.
When compared with previous reports, our case has two special features. First, a perigastric LN involvement is unique. In a recent update that included 99 patients with PDNEC with an unknown primary site [4], the majority of patients had multiple sites of metastasis, and commonly involved sites included the retroperitoneum in 20.2%, peripheral LN in 14.1%, mediastinum in 12.1%, liver in 12.1%, and bone in 11.1%. Accordingly, perigastric LN involvement is unusual for neuroendocrine carcinomas of unknown primary origin. Second, in almost all previous reports, only chemotherapy (usually platinum-based) was administered [4], whereas, given the unusual setting and a tumor limited to a single site, we adopted surgical resection followed by chemotherapy. A small number of previous case reports have also described treatment of PDNEC of unknown primary origin by surgical resection. Hisamori et al. [6] reported two cases of PDNEC of unknown primary origin in an inguinal and a pancreaticoduodenal LN, respectively, which were treated by surgical resection alone [8]. Considering the fact that both patients achieved long-term disease-free survival with LN dissection only, their cases probably could have originated from the affected LN itself. However, in general, PDNEC is highly aggressive, with a high rate of metastases and recurrence [1,2]. In addition, this disease group represents a favorable subset of unknown primary carcinomas that are sensitive to platinum-based combination chemotherapy [1,9,10]. Therefore, aggressive treatment of PDNEC of unknown origin following guidelines similar to those for small cell lung cancer is advised and if possible, a platinum-base adjuvant chemotherapy should be considered.
Neuroendocrine carcinoma of unknown primary origin is very rare, and its diagnosis is difficult. In this unusual setting, because a minority of patients could enjoy long-term and disease-free survival, we recommend treatment of patients with a single or localized PDNEC from an unknown primary site with an aggressive local therapy. In addition to definitive local therapy, the authors believe that these patients should also receive either neoadjuvant or adjuvant chemotherapy.
Go to :
 XML Download
XML Download