

 PDF
PDF Citation
Citation Print
Print


Introduction
c-Met is a member of the sub-family of receptor tyrosine kinases (RTKs) for hepatocyte growth factor (HGF) [1]. It is involved in a large number of cellular processes such as cell proliferation, migration, and invasion [2]. Abnormal c-Met activation is related to cancer development and metastasis and correlates with poor prognosis in a number of human cancers including lung cancer [3]. Likewise, c-Met amplification is known to be involved in the resistance to gefitinib in lung cancer [4]. Given the important role of aberrant HGF/c-Met signaling in lung cancer, c-Met tyrosine kinase inhibitors (TKIs), which are ATP competitive small molecule inhibitors of the catalytic activity of c-Met, have recently been studied in preclinical and clinical models [5]. c-Met TKIs inhibit tumor growth and angiogenesis in lung cancer cells and animal models [6,7]. However, the mechanisms mediating the sensitivity to c-Met TKIs in lung cancer remain unclear.
c-Met activates phosphatidyl-inositol-3' kinase (PI3K), a major intracellular mediator of growth and survival [2], and PI3K can antagonize p53-triggered apoptosis [8]. Crosstalk between the p53 and PI3K pathways occurs at multiple levels via specific downstream regulators such as PTEN and mdm2 [9,10]. In particular, mdm2 is a negative regulator of p53 and mediates degradation of p53 protein [11]. Recently, Moumen et al. [12] reported that c-Met regulates the level of mdm2 via mTOR/Akt signaling in liver development, and that activation of PI3K by c-Met leads to the inhibition of p53-dependent death in liver development. A few studies showed that the loss of p53 function enhances inappropriate c-Met-mediated carcinogenesis [13]. Although c-Met signaling could be related to inactivation of p53 protein in cancer development, the role of p53 in the inhibition of c-Met signaling is not fully understood. In this study, to clarify whether p53 influences cell death through the blocking of c-Met signaling in lung cancer, we examined the possibility of SU11274-induced apoptosis through p53 protein and found that SU11274 indeed caused apoptosis through the regulation of p53 expression in lung cancer cells, as well as in a xenograft model.
Go to : 
Materials and Methods
1. Cell culture and chemicals
A549, NCI-H460, NCI-H1299, and NCI-H358 cells were purchased from the American Type Culture Collection (Rockville, MD). Calu-1 was obtained from Dr. Jong Kuk Park at Korea Institute of Radiological and Medical Sciences (KIRAMS, Seoul, Korea). A549 cells were grown in F12 media (GibcoBRL, Grand Island, NY), whereas NCI-H460, NCI-H358, NCI-H1299, and Calu-1 cells were grown in RPMI1640 media (GibcoBRL) supplemented with 10% fetal bovine serum (FBS; GibcoBRL) and 1% penicillin-streptomycin (GibcoBRL) in a humidified atmosphere with 5% CO2 at 37℃. SU11274 and cycloheximide (CHX) were purchased from CalBiochem (La Jolla, CA), tetracycline was from Amresco (Solon, OH), Z-DEVD fmk was from R&D Systems (Minneapolis, MN), and MG132 was from Sigma (St. Louis, MO).
2. Cell proliferation assay
Cell proliferation was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. 2×103 cells were seeded in 96-well plates and then treated with either 0.1% dimethyl sulfoxide (DMSO) as a diluent control or the indicated concentration of SU11274 in complete medium with 10% FBS for 3 days. MTT solution was added for 4 hours. After the used media containing MTT solution was removed, the formed formazan crystals were dissolved in DMSO. The plates were read at 540 nm in a microplate reader (BioRad, Hercules, CA).
3. Cell counting assay
Cell proliferation was evaluated by the trypan-blue exclusion method. For trypan-blue exclusion experiments, 3×105 cells per well were seeded in 60-mm dishes, and increasing concentrations of SU11274 or an equal volume of diluent (DMSO) were added to complete medium. Cell number was evaluated at the indicated time points using a hematocytometer. Proliferation was expressed as a percentage relative to day 0.
4. Colony forming assay
Four hundred cells were seeded in 6-well plates. SU11274 was applied at the indicated concentration for 72 hours. The cells were cultured in fresh media for 2-3 weeks. When colonies became visible, those larger than 0.5 mm in diameter were stained with 1% crystal violet in methanol and counted using a colony counter (Imaging Products, Chantilly, VA).
5. Cell cycle analysis
Cell cycle was analyzed by propidium iodide (PI) staining. Briefly, both adherent and non-adherent cells were harvested and fixed with 70% ethanol. Fixed cells were incubated with 50 mg/mL of PI and 100 mg/mL of RNase at 37℃ for 30 minutes and then analyzed by flow cytometry. The data were analyzed with CellQuest software (BD Bioscience, San Jose, CA).
6. Apoptosis assay
Both adherent and non-adherent cells were harvested and fixed with 1% paraformaldehyde. Apoptotic cells were stained using the APO-BRDU kit (Phoenix Flow Systems, San Diego, CA) according to the manufacturer's instructions. Cells treated with DMSO were used as the negative control, and HL-60 leukemic cells treated with camptothecin provided with the kit were used as the positive control. Apoptotic cells were evaluated on a BD FACScalibur (Becton Dickinson, Franklin Lakes, NJ). The data were analyzed with CellQuest software.
7. Western blot analysis
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and harvested. Harvested cells were lysed in lysis buffer (10 mM Tris-Cl [pH 7.4], 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.2% Triton X-100) containing protease inhibitors and phosphatase inhibitor for 60 minutes at 4℃. Cell lysates were separated by 8-12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (Amersham Co., Arlington Heights, IL). These membranes were incubated overnight with primary antibodies raised against phospho-c-Met, phospho-Akt, Akt, PUMA, poly(ADP-ribose) polymerase (PARP; Cell Signaling Technology, Danvers, MA), c-Met, p53, mdm2, Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), Bax (Daco, Glostrup, Denmark) and β-actin (Sigma-Aldrich) at 4℃. Membranes were then washed with Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) and incubated with appropriate secondary antibodies for l hour. After washing in TBS-T, an ECL kit (Amersham Co.) was used to detect the target protein.
8. Immunofluorescence staining
2×104 cells were seeded on coverslips, fixed for 15 minutes in 4% paraformaldehyde on ice, washed in PBS containing 0.1% Tween 20 (PBS-T), and permeabilized with 0.1% Triton X-100 for 30 minutes. Nonspecific binding of antibodies was blocked by incubating the cells in PBS containing 3% bovine serum albumin and 0.1% Tween 20 at room temperature for 1 hour. The cells were incubated overnight with primary antibody (1 : 200) at 4℃ and appropriate secondary antibodies (1 : 500) were applied at room temperature for 30 minutes. To observe the nuclei, DAPI staining was performed for 15 minutes. Between the steps, cells were washed 3 times with PBS-T for 10 minutes at room temperature. Stained sections were mounted and observed under a fluorescence microscope (Olympus, Tokyo, Japan).
9. Plasmid construction and gene transfection
The human p53 cDNA coding region was cloned into pcDNA3 (Invitrogen Co., Carlsbad, CA). The T-REx complete kit, including pcDNA6/TR, and pcDNA4/TO-p53 plasmids (Invitrogen Co.), was kindly provided by Jin Kyung Rho in KIRAMS (Seoul, Korea). Calu-1 cells with null-type p53 were transfected with two expression vector systems such as pcDNA3, pcDNA3-p53 and pcDNA6/TR, pcDNA6/TR-pcDNA4/TO-p53. Transfections were performed with Lipofectamine 2000 (Invitrogen Co.) according to the protocol recommended by the manufacturer. To generate stable transfectants, media on pcDNA3 and pcDNA3-p53-transfected Calu-1 cells were refreshed every 2 days with 1 mg/mL G418-containing medium for 4-6 weeks. For tetracycline-p53 stable transfectants, the pcDNA4/To-p53 plasmid was cotransfected into Calu-1 cells with pcDNA6/TR and TR, and the media on TO-p53-transfected cells were changed every 2 days with fresh 10 µg/mL blasticidin-containing medium and 800 µg/mL zeocin-10 µg/mL blasticidin-containing medium, respectively, for 4-6 weeks. After a colony was picked, the expression of p53 was confirmed by Western blot analysis.
10. siRNA transfection
Specific siRNAs targeting human p53 mRNA were purchased from Santa Cruz Biotechnology Inc. The p53 siRNA duplexes were transiently transfected using Lipofectamine 2000 (Invitrogen Co.), according to the manufacturer's instructions. Control transfection using scrambled siRNA was performed in parallel using control siRNA (Santa Cruz Biotechnology Inc.). Transfected cells were used for Western blot and flow cytometric analyses.
11. In vivo study
Six-week-old female BALB/c nude mice were purchased from Charles River Laboratories (Wilmington, MA). A549 xenografts were established by intradermally injecting 7×106 cells into the flank of nude mice. Briefly, cells were suspended in 100 µL of corn oil and subcutaneously injected into the flank of each mouse. Tumors were grown until they reached a median size of 200-300 mm3 (35 days). Animals were randomly divided into two groups (n=3-4) and treated with SU11274 (100 µg per xenograft). Tumor sizes were measured in two dimensions using a caliper, and tumor volumes were calculated by the formula of short axis2×long axis/2. At the end of the treatment period, animals were sacrificed and tumors were removed. All animal experiments were approved by local ethics authorities.
12. Ex vivo analysis of tumor material
Tissues were fixed in PBS-buffered formalin (10%) and subsequently embedded in paraffin. For immunohistochemistry, dewaxed 4-µm-thick tissue sections were processed in citrate buffer (pH 6) with retrieval cycles of 5 minutes in a microwave oven. After blocking with 0.1% Triton X-100 and 10% normal goat serum in PBS for 1 hour at room temperature, the sections were incubated overnight with primary antibodies in the same solution at room temperature. p53 antibody (Daco) was added at a dilution of 1 : 200. Sections were further processed with biotinylated secondary antibodies (1 : 300) and the avidin-biotin-peroxidase complex, and finally visualized with 3,3'-diaminobenzidine (Roche, Mannheim, Germany). Alternatively, sections were processed for apoptosis analysis by terminal deoxyribonucleotide transferase-mediated nick-end labeling (TUNEL) staining (Apoptag, Chemicon International, Temecula, CA) according to the manufacturer's instructions.
13. Statistical analysis
All in vitro experiments were repeated at least three times. Data were calculated with Microsoft Excel software (Microsoft Corporation, Seattle, WA) and MINITAB 14 software (Minitab Inc., State College, PA). Differences between experimental groups were determined by Student's t-test with Microsoft Excel software. Every error bar indicates standard error, and p-values of <0.05 were considered statistically significant.
Go to :
Results
1. SU11274 shows anticancer effect in lung cancer cells with wild-type p53
To examine whether SU1124 induced different cell death depending on the p53 status, we used various lung cancer cells with different p53 gene statuses. As shown in Fig. 1A, SU11274 significantly suppressed cell viability in lung cancer cells with wild-type p53. Conversely, it did not inhibit cell proliferation in null-type p53 cells. Furthermore, SU11274 increased the level of p53 protein in wild-type p53 groups (Fig. 1B). For further study, we used A549 and Calu-1 cell lines, which are wild-type p53 and null-type p53 lung cancer cells, respectively. These cells have higher expression of endogenous c-Met than other cells (data not shown) and wild-type PTEN. First, we examined whether HGF stimulation induced the activation of MET/Akt/Erk signaling in A549 and Calu-1 cells. The basal level of MET protein was higher in Calu-1 cells than A549 cells (data not shown). The phosphorylation of MET, Akt, and Erk protein was stimulated by HGF (100 ng/mL) in both cell lines, indicating that A549 and Calu-1 cells have intact MET/Akt/Erk signaling pathway. To further confirm the effect of SU11274 on cell growth inhibition, A549 and Calu-1 cells were treated with SU11274 of various concentrations and for various periods. A549 cells were more sensitive to SU11274 than Calu-1 cells (Fig. 1C). The survival fraction after SU11274 treatment was effectively decreased in A549 cells compared with that in Calu-1 cells (Fig. 1D). Next, whether SU11274 regulated the phosphorylation of MET and downstream mediators such as Akt and Erk was investigated (Fig. 1E). SU11274 effectively suppressed the phosphorylation of MET, Akt, and Erk proteins in a time-dependent manner in Calu-1 cells. Although SU11274 increased the activation of MET signaling in A549 cells early, it also inhibited the phosphorylation of MET, Akt, and Erk proteins late. To identify the mechanism of growth inhibition by SU11274, the change of cell cycle and apoptosis in those cells were analyzed. SU11274 induced G2/M arrest in A549 cells at 6 hours after treatment. After 72 hours of treatment, SU11274 induced sub-G1 in ~35% of A549 cells, whereas the sub-G1 fraction was not increased in Calu-1 cells (Fig. 2A). In addition, apoptosis analysis (Fig. 2B) showed that SU11274 induced apoptosis in ~50% of A549 cells, but not in Calu-1 cells. As shown in Fig. 2C, A549 cells after SU11274 treatment clearly showed a significant increase of cleaved PARP expression, while the levels of pro-caspase 9, 8, and 3 expression were markedly reduced. On the other hand, cleaved PARP and pro-caspases in Calu-1 cells were not changed after SU11274 treatment.
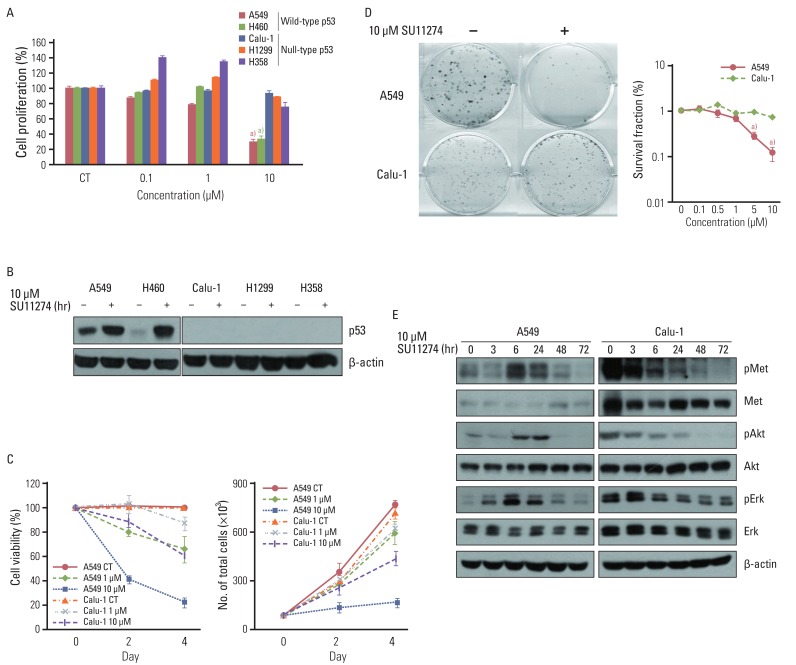 | Fig. 1Effect of SU11274 on growth inhibition in lung cancer cells. (A) The result of MTT assay in lung cancer cells with different p53 statuses treated with the indicated concentrations of SU11274 for 72 hr. Columns, mean value of six identical wells of a single representative experiment; Bars, standard error. a)p<0.001 for comparisons between SU11274-treated and untreated cells. (B) The expression of p53 protein in lung cancer cells. Cells were treated with 10 µM SU11274 for 24 hr, and cell extracts were immunoblotted using p53 antibody. β-actin serves as a loading control. (C) Results of cell counting in A549 and Calu-1 cells treated with 1 and 10 µM SU11274 for 48 hr and 96 hr. Points, mean value of three experiments; bar, standard error, this data presented p < 0.05; CT, control. (D) Results of colony-forming assay in A549 and Calu-1 cells treated with indicated concentrations of SU11274 for ~3 weeks. The surviving fraction (SF) was calculated from the following formula: SF=number of colonies formed/number of cells seeded×plating efficiency of control group. Points, mean values of three experiments; bar, standard error. a)p<0.001. (E) A549 and Calu-1 cells were treated with 10 µM SU11274 for indicated time. Whole cell lysates were prepared and separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblotted for MET, Akt, Erk and their phosphorylated forms. β-actin was used as a loading control.
|
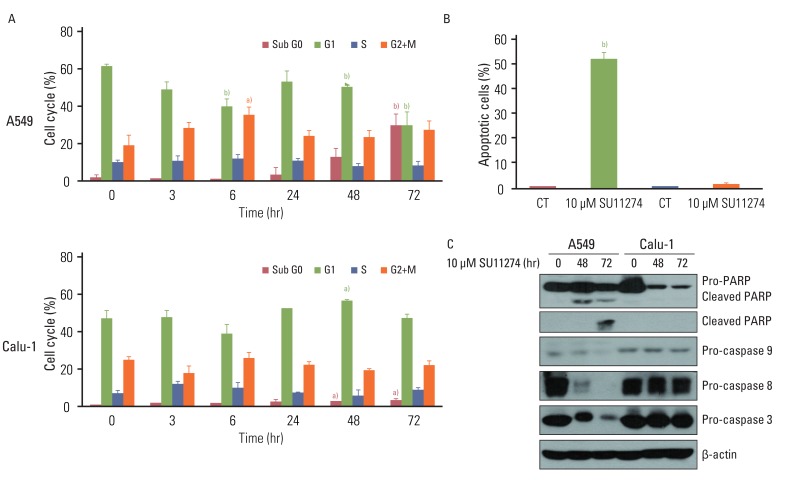 | Fig. 2Effects of SU11274 on apoptosis in A549 and Calu-1 cells. (A) Results of cell cycle analysis using propidium iodide staining in SU11274-treated A549 and Calu-1 cells. Cells were treated with 10 µM SU11274 for indicated period of time, and both adherent and floating cells were harvested for analysis of cell cycle distribution. Columns, mean value of three experiments; bar, standard error. a)p<0.05, b)p<0.001. (B) The results of apoptosis in A549 and Calu-1 cells assessed using APO-BRDU kit. A549 and Calu-1 cells were treated with 10 µM SU11274 for 72 hr, and both adherent and floating cells were harvested. Columns, mean value of three identical experiments; bars, standard error; CT, control. b)p<0.001 for comparisons between SU11274-treated and untreated cells. (C) Expression of poly(ADP-ribose) polymerase (PARP), caspase 9, caspase 8, and caspase 3 in A549 and Calu-1 cells. Cells were treated with 10 µM SU11274 for 48 and 72 hr. Both adherent and floating cells were harvested.
|
2. SU11274 increases p53 protein in A549 cells with wild-type p53
To elucidate the possible role of p53 in SU11274-mediated apoptosis, we examined whether SU11274 regulated the expression of p53 protein. As shown in Fig. 3A and 3B, SU11274 treatment in A549 cells induced p53 expression in a time-dependent manner without changing the cellular localization of p53 protein. The increase of p53 protein level is due to stabilization of p53 protein against degradation. As shown in Fig. 3C, the level of p53 protein in SU11274-treated A549 cells was not changed in the presence of CHX (protein synthesis inhibitor), whereas the level of p53 protein was markedly reduced in SU11274-untreated cells (the half-life of p53 protein was ~60 minutes). Furthermore, SU11274 stabilized p53 protein for 6 hours in A549 cells when they were treated with MG132 (proteasome inhibitor) and SU11274, compared with MG132 only-treated A549 cells (Fig. 3D). These data indicated that SU11274 increased the stability of p53 protein in A549 cells. c-Met regulates the activity of Akt protein, and activated Akt modulates mdm2 protein, a p53 negative regulator [12]. Therefore, to elucidate the mechanism involved in SU11274-mediated increase of p53 protein, we investigated whether SU11274 regulated c-Met, Akt and mdm2 proteins in A549 cells. Fig. 3E shows that SU11274 inhibited the activity of c-Met and Akt proteins and the level of mdm2 protein. The above findings indicate that SU11274 decreased mdm2 protein through blockade of c-Met/Akt signaling and induced p53 protein through enhancement of p53 stability.
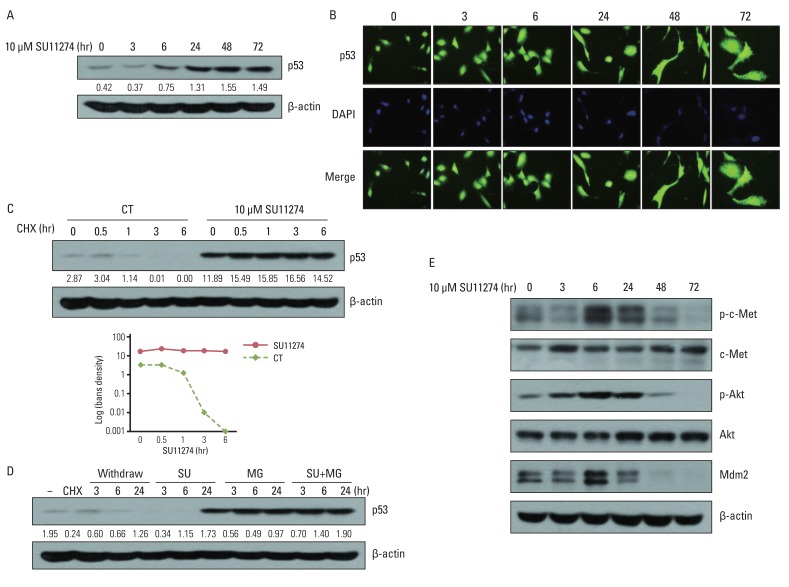 | Fig. 3Effects of SU11274 on p53 protein in A549 cells. (A) The expression of p53 protein in SU11274-treated A549 cells. Cells were treated with SU11274 for the indicated periods of time. Whole cell lysates were prepared and separated by 10% sodium dodecyl sulfatepolyacrylamide gel electrophoresis, and immunoblotted using p53 antibodies. β-actin serves as a loading control. Bands were quantified by densitometry. (B) The effects of SU11274 on cellular localization of p53 protein in A549 cells. SU11274-treated A549 cells immunostained with anti-p53 antibody (green fluorescence) and DAPI (blue fluorescence) in a time-dependent manner, and analyzed by fluorescence microscopy to determine the cellular localization of p53. (C) p53 stability in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274 for 24 hr, followed by 10 µg/mL cycloheximide (CHX) for the indicated periods of time. CT, control. (D) p53 increase in SU11274-treated A549 cells. Cells were treated with 10 µg/mL CHX for 6 hr, followed by change of new growing medium only and new growing medium containing 10 µM SU11274, 10 µM MG132, or both for indicated times. Each band was quantified by densitometry. SU, SU11274; MG, MG132. (E) The expression of phospho-c-Met (p-c-Met), c-Met, phospho-Akt (p-Akt), Akt, and mdm2 protein. Cells were treated with 10 µM SU11274 for the indicated time.
|
3. Increased p53 protein by SU11274 induces apoptosis through the regulation of Bax, PUMA, and Bcl-2 in A549 cells
To further investigate the mechanisms responsible for increased p53 protein-induced apoptosis in A549 cells, we examined whether increased p53 regulated apoptosis-related proteins (PUMA, Bax, and Bcl-2). As shown in Fig. 4A, SU11274 increased the levels of Bax and PUMA expression and decreased the level of Bcl-2 expression in A549 cells in a time-dependent manner. Next, we examined whether activated caspase 3 was necessary for SU11274-induced apoptosis. We used Z-DEVD-fmk to inhibit the activation of caspase 3 and found that the caspase 3 inhibitor significantly suppressed the SU11274-induced apoptosis in A549 cells (Fig. 4B).
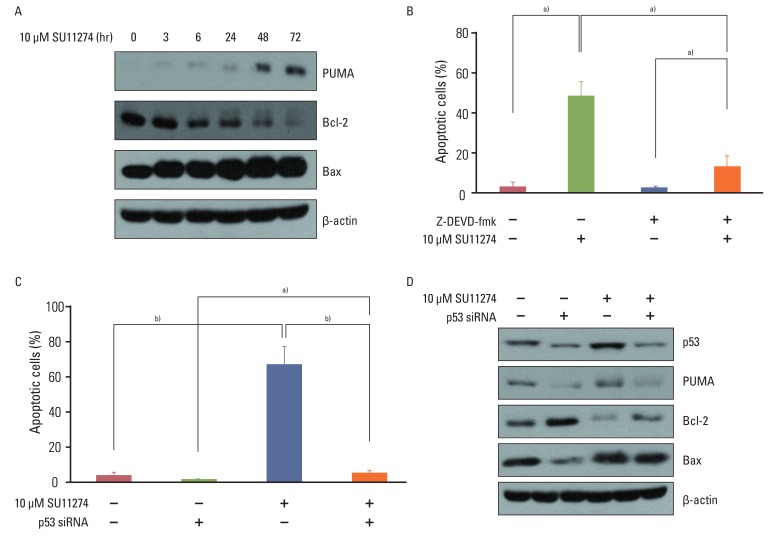 | Fig. 4The role of p53 in SU11274-mediated apoptosis in A549 cells. (A) Expression of Bax, PUMA, and Bcl-2 in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274 for the indicated times. (B) Effects of caspase 3 on apoptosis in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274, 2 µM Z-DEVD-fmk (caspase 3 inhibitor), or both for 72 hr. Both adherent and floating cells were harvested and evaluated by the APO-BRDU kit. Columns, mean value of three experiments; bar, standard error. a)p<0.05. (C) Effects of p53 siRNA on apoptosis in SU11274-induced apoptosis using APO-BRDU kit. Cells were transiently transfected with scramble siRNA or p53 siRNA. After 24 hr, cells were treated with 10 µM SU11274 for 72 hr. Both adherent and non-adherent cells were harvested. Columns, mean value of three experiments; bar, standard error. a)p<0.05, b)p<0.005. (D) Expression of p53, Bax, PUMA, and Bcl-2 protein in p53 siRNA-transfected A549 cells after SU11274 treatment.
|
4. p53 plays a critical role in SU11274-mediated apoptosis in lung cancer cells
To confirm that SU11274-mediated apoptosis depends on p53, we analyzed whether the inhibition of p53 protein by p53 siRNA duplexes influenced the sensitivity to SU11274. As shown in Fig. 4C, the blocking of p53 protein by p53 siRNA duplexes significantly suppressed SU11274-mediated apoptosis. The levels of Bax and PUMA expression were decreased when SU11274-treated A549 cells were transfected with p53 siRNA compared with scrambled siRNA. Conversely, the level of Bcl-2 expression was increased in the presence of p53 siRNA (Fig. 4D). Next, to clarify whether exogenous p53 induced SU11274-mediated apoptosis, Calu-1 cells with p53 null-type were investigated. For the expression of p53, we used two systems including the full-length p53 pcDNA3 expression vector and T-Rex complete kit. Western blot analysis confirmed the induction of p53 by tetracycline treatment in Calu-1 cells (Fig. 5A). We then tested whether SU11274 caused apoptosis after the introduction of p53 into Calu-1 cells. As shown in Fig. 5B and 5C, SU11274 induced more than 40% apoptosis in all p53 transfectants. In particular, the level of p53 expression in Calu-1 cells with exogenous p53 was similarly increased to that observed in SU11274-treated A549 cells (Fig. 5D). Likewise, SU11274 induced up-regulation of Bax and PUMA expression and down-regulation of Bcl-2 expression in p53 transfectants compared with that in empty transfectants.
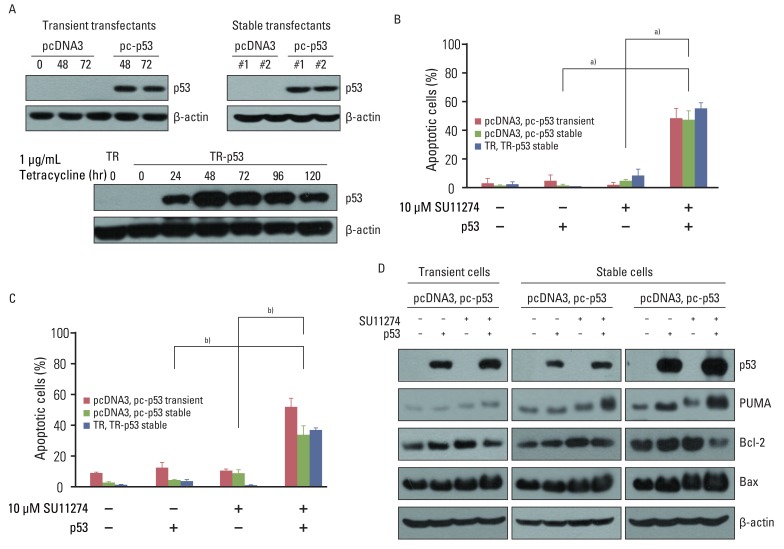 | Fig. 5Effects of exogenous p53 protein on SU11274-mediated apoptosis. (A) Expression of p53 protein in p53 transfectants. Cells were transfected with pcDNA3, pcDNA3-p53 (pc-p53) expression vectors, pcDNA6/TR (TR), or pcDNA6/TR-pcDNA4/TO-p53 (TR-p53) expression vectors. To prepare transient transfectants, cells were transfected with pcDNA3 and pc-p53. After 48 and 72 hr, cells were harvested and immunoblotted using p53 antibodies. Stable transfectants, described in Materials and Methods, were harvested and analyzed by immunoblotting. One µg/mL Tet analogue tetracycline was applied to TR and TR-p53 stable transfectants for 24 hr and the cells were harvested at the indicated times. Cell extracts were analyzed by immunoblotting using p53 antibody. β-actin was used as a loading control. (B) Effects of exogenous p53 protein on SU11274-induced apoptosis in empty vector and p53 transfectants. TR and TR-p53 stable transfectants were pretreated with 1 µg Tet analogue tetracycline for 24 hr and total cells were treated with 10 µM SU11274 for 72 hr. Both adherent and non-adherent cells were harvested. Columns, mean value of three experiments; bar, standard error. a)p<0.005. (C) Result of cell cycle analysis using propidium iodide staining in p53 transfected Calu-1 cell after SU11274 treatment. Cells were transiently transfected with pcDNA3 and pc-p53 DNA. After 24 hr, cells were treated with 10 µM SU11274 for 72 hr. Stable p53 transfectants were also treated with 10 µM SU11274 for 72 hr. Both adherent and floating cells were harvested for analysis of cell cycle distribution. Columns, mean value of three experiments; bar, standard error. b)p<0.001. (D) Expression of p53, Bax, PUMA, and Bcl-2 proteins in empty vector and p53 transfectants. Cells were treated with 10 µM SU11274 for 72 hr.
|
5. SU11274 induced apoptosis through the increase of p53 protein in tumor xenograft models
To test whether SU11274 affects tumor growth in an in vivo model, a human A549 xenograft was used. SU11274 (100 µg per xenograft) was given when the tumor began to be visible (~250 mm3) and was applied daily or twice weekly at the indicated times. As shown in Fig. 6A, SU11274 significantly suppressed the xenograft growth in daily and twice-weekly treatment groups compared with that in the DMSO-alone group. Similarly, SU11274 caused marked apoptosis in the SU11274-treated A549 xenograft model (Fig. 6B). Next, we sought to confirm whether SU11274 accumulates p53 protein in vivo; we therefore evaluated the level of p53 protein in tumor tissues from the A549 xenograft model. As shown in Fig. 6C, SU11274-treated tumor tissues showed approximately 3- and 5-fold increases of p53 protein in the daily and twice weekly-treated groups, respectively. Furthermore, immunohistochemical analysis revealed significantly increased levels of p53 expression in the SU11274-treated tumor tissues of the two models (Fig. 6D).
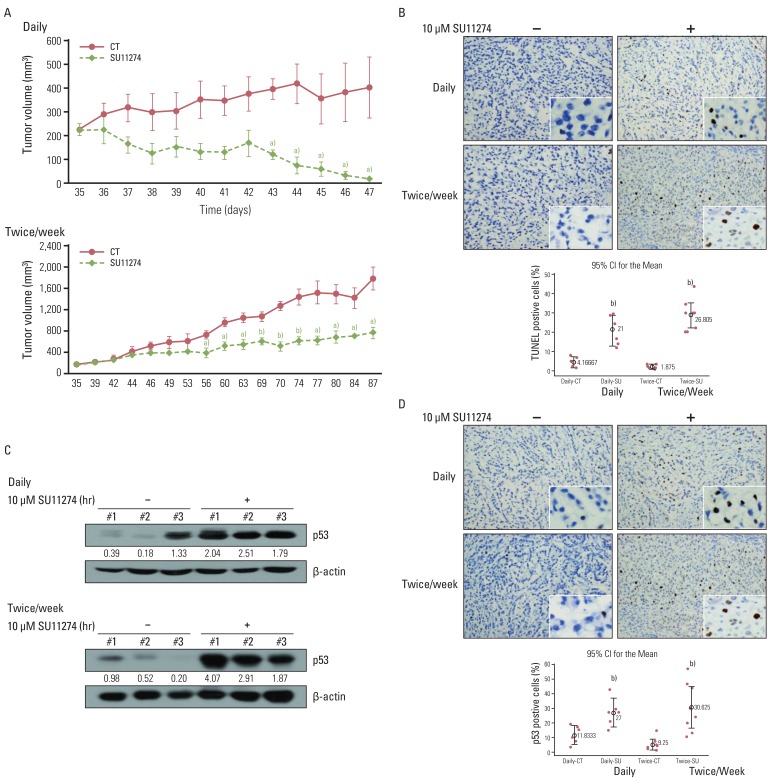 | Fig. 6Effects of SU11274 on tumor growth in A549 xenograft model. (A) Change of tumor volume after treatment with SU11274 daily and twice weekly for the indicated periods in the xenograft model. SU11274 (100 µg per xenograft) was administered on two schedules through intratumoral injections; it was administered daily for 13 days or twice weekly for 53 days. The tumor volume was recorded at the indicated times. n=7 (dimethyl sulfoxide [DMSO]-treated group [n=3] and SU11274-treated group [n=4]). Animal experiments were performed done twice independently. Points, mean value of three or four identical mice; bars, standard error. a)p<0.05, b)p<0.001. (B) Tumors were harvested on the last day, and apoptosis was assessed using the terminal deoxyribonucleotide transferase-mediated nick-end labeling (TUNEL) assay. Cells stained brown indicate apoptotic cells. CT, control; SU, SU11274. (C) Expression of p53 in tumor tissue harvested from A549 xenograft. Tumor tissue was lysed with homogenizer, resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblotted using p53 antibodies. Bands were quantified by densitometry. (D) Harvested tumor tissue was immunohistochemically examined with anti-p53 antibody. Brown color indicates the expression of p53 protein. The percentage of positive cells was determined from the following formula: The percentage of terminal deoxyribonucleotide transferase-mediated nick-end labeling/p53-positive cells=positive staining cells/total cells×100. Box, interquartile range of six-eight identical mice;⌖, mean value of six or eight identical mice. b)p<0.001 for comparisons between SU11274-treated and untreated mice.
|
Go to :
Discussion
Current therapeutic strategies for lung cancer have a limited effect in the treatment of patients. Therefore, alternative novel therapeutic targets are needed to improve the treatment outcome for lung cancer. c-Met has recently been identified as an attractive anti-neoplastic therapeutic target [14,15]. c-Met targeted agents that block the activation of c-Met signaling have been extensively studied in a preclinical model of lung cancer. c-Met TKIs suppressed the cell viability in c-Met expressing non-small cell lung carcinoma (NSCLC) through abrogation of c-Met phosphorylation and its downstream signaling [16], and short hairpin RNA-mediated c-Met knockdown induced significant growth inhibition in NSCLC cells with c-Met amplification [17]. Consistent with previous reports, we found that SU11274 effectively caused cell death and apoptosis in lung cancer cells harboring wild-type p53. Furthermore, SU11274 continuously suppressed tumor growth in the xenograft model for approximately 2 months, suggesting that c-Met targeted drugs are a potential treatment strategy for improving the survival rate in lung cancer patients.
The amount of p53 protein is mainly regulated by mdm2, a negative regulator [18]. Mdm2 protein binds to p53 at its NH2-terminus leading to nuclear export and promoting rapid degradation of p53 through the ubiquitin proteasome-mediated pathway [19]. An increase of p53 levels results in large part from stabilization of p53 protein against degradation by inhibiting mdm2 binding [20]. In the present study, we found that SU11274 increased the level of p53 protein in A549 cells and in tumor tissue from SU11274-treated mice. On the other hand, mdm2 protein was decreased after SU11274 treatment. Nevertheless, cellular relocalization of p53 and mdm2 protein from cytoplasm to nucleus did not occur (result of mdm2; data not shown). Furthermore, SU11274 continuously maintained the stability of the protein for more than 6 h. Conversely, degradation of p53 protein under normal conditions occurred in less than 1 hour. Thus, SU11274 increased the level of p53 protein via induction of protein stability, suggesting that blocking c-Met signaling stabilized p53 protein by decreasing mdm2 protein. Unfortunately, however, we did not show the exact interaction between mdm2 and p53 protein under abrogation of c-Met signaling. Further detailed studies are needed to clarify this mechanism.
Here, we showed that SU11274 suppressed cell proliferation and tumor growth in A549 cells via inhibition of c-Met signaling. SU11274-treated A549 cells decreased the activity of c-Met, Akt, and Erk after 24 hours of treatment (result of Erk; data not shown), in good agreement with previous studies which showed that it suppressed the cell viability and motility via inactivation of c-Met, Akt, and Erk proteins in NSCLC cells and mouse models [21,22]. Until now, however, the mechanisms involved in the inhibition of c-Met singling have remained obscure. Several studies have suggested that PI3K/Akt signaling regulates the mdm2-p53 pathway through RTK. Insulin-like growth factor receptor inhibition increases p53 stability through modulation of a p53-mdm2 feedback loop [23], and Her2 induces p53 degradation via Akt-mediated mdm2 phosphorylation [24]. In the present study, we found that SU11274 reduced the level of mdm2 protein, accompanied by inactivation of c-Met and Akt protein, thus demonstrating that inactivation of c-Met signaling by SU11274 regulates the level of p53 protein through modulation of mdm2 expression.
Loss of p53 function through gene mutation and deletion contributes to chemotherapy resistance [25], implying that p53 plays a critical role in the sensitivity to drugs. Therefore, we hypothesized that the sensitivity to SU11274 is dependent on the p53 gene status in lung cancer; indeed, we found that SU111274 remarkably induced apoptosis in lung cancer cells with wild-type p53 and in A549 xenograft models compared with lung cancer cells with p53 null-type. Introduction of p53 into Calu-1 cells induced SU11274-mediated apoptosis. Conversely, the loss of p53 function by p53 siRNA significantly reduced apoptosis in SU11274-treated A549 cells. To the best of our knowledge, this is the first study to show the critical role of p53 protein in SU11274-mediated apoptosis. These results suggest that responsiveness to SU11274 is a characteristic of distinct subgroups of lung patients with wild-type p53.
Taken together, SU11274 significantly induced apoptosis in lung cancer cells and in a xenograft model with wild-type p53. SU11274 accumulated p53 protein through increased p53 stability. Increased p53 protein caused apoptosis via regulation of PUMA, Bax, and Bcl-2, and activation of the caspase family. In p53 knock-out and knock-in systems, we confirmed that SU11274 caused apoptosis through the p53-mediated apoptotic pathway. In addition, SU11274 effectively decrease tumor volume and induced apoptosis via increased p53 protein expression in the A549 xenograft model.
Go to :
 XML Download
XML Download