

 PDF
PDF Citation
Citation Print
Print


Introduction
Erdheim-Chester disease (ECD) is a rare non-Langerhans-cell histiocytosis of unknown etiology. Since it was first described by Erdheim and Chester in 1930 [1], approximately 400 or more cases of ECD have been reported in the literature. Since the first case of ECD was described in 1999, 10 cases have been reported in Korea [2]. Age at onset is 40-60 years. The disease is characterized by infiltration of lipid-laden histiocyte-producing xanthogranulomas in long tubular bones, leading to osteosclerosis. Xanthogranulomatous tissues can also be found in skin, lung, heart, kidney, retroperitoneal space, orbit, and pituitary gland. Bone pain is the most common symptom, and bilateral osteosclerosis of long bones, excluding the epiphysis, is regarded as a typical radiological finding [3]. Results of histopathological analysis show large, foamy or lipid-laden histiocytes with eosinophilic cytoplasm and/or pathognomonic Touton-like giant cells [4]. ECD is usually a progressive disease, with death from cardiac, pulmonary, or renal involvement occurring in approximately 50% of cases. We report on a patient with an indolent course of the disease, who presented with asymptomatic renal involvement followed by mild limb pain.
Case Report
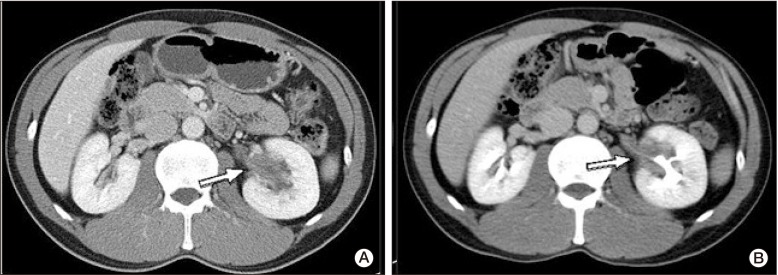
A 41-year-old man presented with left hydronephrosis, detected incidentally during a regular checkup in July 2008. A computed tomography (CT) scan of his abdomen showed an infiltrative homogeneous mass involving the left renal sinus, with low attenuation on CT (Fig. 1A). Results of a ureteroscopic biopsy revealed nonspecific inflammation. However, result of urine analysis and microscopic examination showed no renal dysfunction or abnormality. Until July 2010, follow-up CT scans showed no significant change in perirenal infiltration (Fig. 1B).
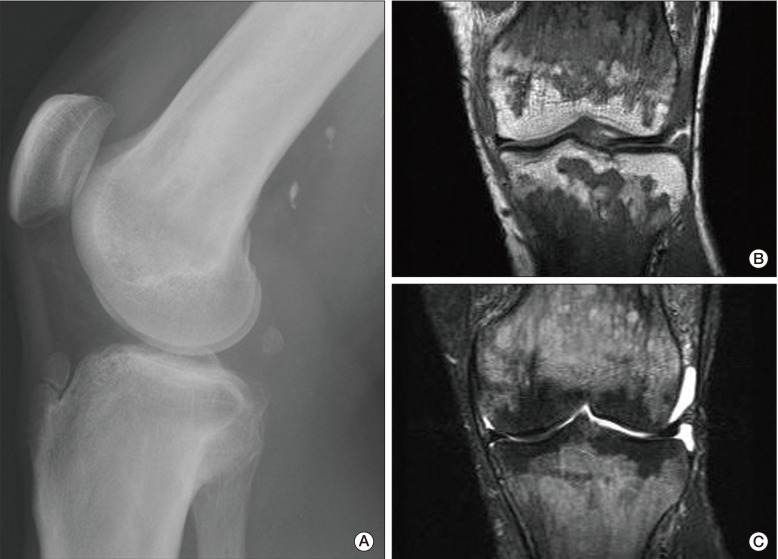
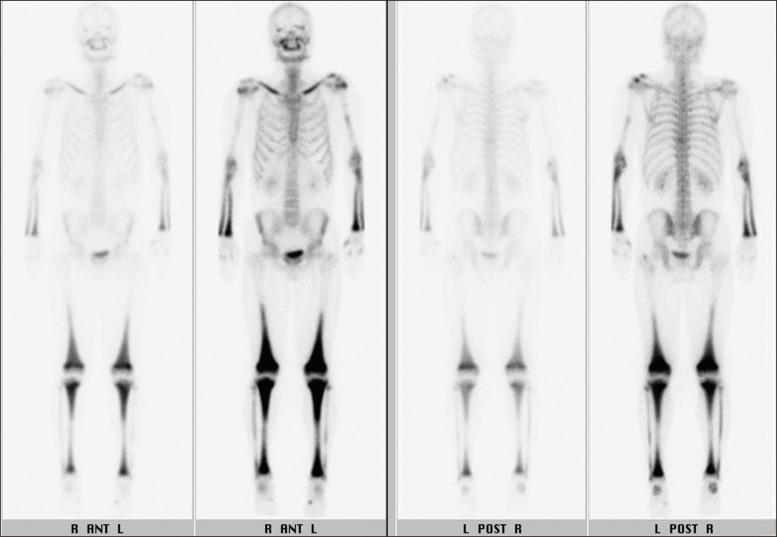
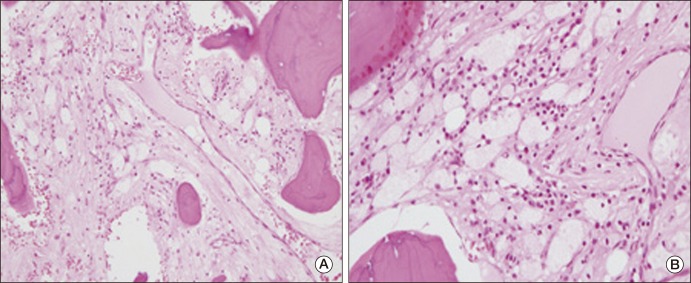
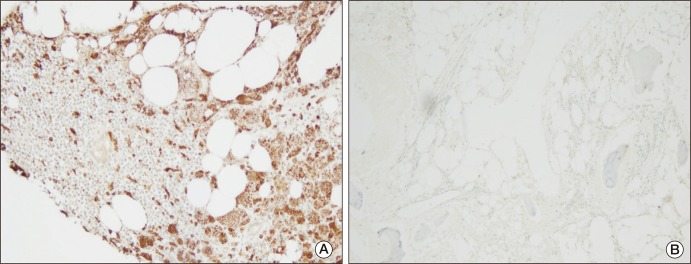
In March 2010, the patient was admitted to the hospital complaining of bone pain that had lasted for 10 months, with no significant history of trauma. He reported mild pain in both knees, which gradually extended to both ankles and both upper limbs. However, the pain he described had not affected his daily life. He showed no sign of fever or weight loss. His erythrocyte sedimentation rate was 23 mm/hr and his C-reactive protein was 3.09 mg/dL. Results of urine analysis, complete blood count, renal and liver function tests, and lipid profiles were normal. Plain radiography of the left knee (Fig. 2A) showed heterogeneous patchy medullary sclerosis and coarse trabeculae and osteopenia of the covered femur and tibia. Symmetric and diffuse sclerotic changes were observed on gadolinium-enhanced magnetic resonance imaging (MRI) of his knees and ankles. Involvement of mainly the meta- and diaphyses, with relative epiphyseal sparing, was observed (Fig. 2B and C). Diffuse and symmetric uptake in the bilateral tibiae, fibulae, distal femora, radii, ulnae, humeri, and clavicles was observed by whole-body bone scintigraphy (Fig. 3). MRI of the brain and CT scan of the chest showed no abnormalities. Biopsy of the proximal tibia revealed sclerotic bony trabeculae, with lipid-laden histiocytic and lymphocytic infiltration (Fig. 4). As a result of immunohistochemical staining, lipid-laden histiocytes were found to be positive for CD68 (Fig. 5A) and negative for S-100 and CD1a (Fig. 5B). Results of a bone marrow examination also revealed two focal lymphohistiocytic infiltrations, which were found to be positive for CD68 and negative for CD1a. Bone marrow examination showed no atypical cells. Bone pain described by the patient did not require intervention; therefore, a wait-and-see policy was adopted in the outpatient clinic.
Discussion
Occurrence of ECD is extremely rare and diagnosis is difficult; therefore, clinical suspicion is an important factor in its diagnosis. Final diagnosis is based on clinical suspicion, classic radiographic findings in long bones, and biopsy, with histological confirmation.
In diagnosis, ECD must be distinguished from other granulomatous disorders. Chronic recurrent multifocal osteomyelitis (CRMO) is of particular concern. CRMO was initially described as a symmetric osteomyelitis; however, since no causative organism has been identified, it is now considered a noninfectious inflammatory disease. The clinical course of CRMO is self limited. CRMO involves the mandible and spine and lesions are associated with local inflammation and prominent periosteal reaction. When involvement of multiple bones is detected, evaluation for tumors such as mastocytosis, myelofibrosis, lymphoma, and osteosclerotic myeloma should be performed. In these tumorous conditions, the presence of many systemic manifestations is observed more often, according to the situation [5].
Morphologically and immunohistochemically, ECD appears to be a member of the juvenile xanthogranuloma (JXG) group. JXG cells express factor XIIIa, CD68, and are negative for CD1a and Lagenrin. S-100 positivity is reported in less than 20% of cases. However, none of these markers is specific for JXG. Involvement of bilateral symmetric long bones in association with extraskeletal organ involvement is a characteristic feature of ECD. ECD occurs mainly in middle aged adults, while other systemic JXG occurs mainly in young children [6].
In this patient, an incidentally detected perirenal infiltrative mass was the first manifestation of ECD. Bone lesions were detected after a two-year asymptomatic period. Perirenal infiltration is a well-known extraskeletal finding of ECD. Veyssier-Belot et al. [3] reported renal and retroperitoneal involvement in 30% of patients. Hydronephosis, an enlarged kidney, and retroperitoneal infiltration surrounding the abdominal aorta and kidneys were observed on abdominal CT in cases of renal and retroperitoneal involvement of ECD. This involvement was usually asymptomatic and CT scan revealed hypoattenuated homogenous tissue infiltration [3]. Although CT images of the current case showed only mild hydronephrosis, the nature of the tissue surrounding the left kidney and ureter was consistent with that of retroperitoneal involvement of ECD; the present case demonstrated typical bone involvement of ECD. Therefore, despite the lack of pathologic confirmation, we suggested that the perirenal infiltrative mass could be considered renal involvement of ECD.
The clinical course of ECD varies. According to the extent of organ involvement, patients present with a range of symptoms extending from a focal asymptomatic state to multisystemic, fatal, infiltrative disease. Primary causes of death include cardiomyopathy, interstitial lung disease, and renal failure. Unfortunately, approximately 60% of patients died of ECD within three years from initial presentation [3]. No treatment for ECD has yet been established; however, treatment of ECD patients can include steroids, cytotoxic agents [3], bisphosphonate [7], and autologous stem cell transplantation [8]. Interferon-α (IFN-α) as a valuable therapy for treatment of patients with ECD, with tolerable toxicity [9-11], and long-lasting responses (3-4 years), has recently been reported.
Renal failure can be attributed to obstructive uropathy in association with retroperitoneal fibrosis (79%) or renal histiocytic infiltration (21%) [12]. However, renal lesions are not responsive to systemic immunosuppressive agents, whereas bone pain and general performance status show improvement with treatment. Most patients with renal involvement die within five years of diagnosis [13]. IFN-α is the only effective agent known to stabilize retroperitoneal fibrosis and cause its regression.
Efficacy of IFN-α varies according to the site of involvement. Treatment with IFN-α appears to be effective in patients with renal involvement, whereas its failure has been reported in patients with severe multisystemic involvement that includes the central nervous system and cardiovascular system [9]. Early therapeutic intervention with INF-α appears to be effective in preventing progression of cardiovascular and renal involvement [11]. Imatinib mesylate has been suggested by some investigators as an experimental alternative therapeutic regimen after failure of INF-α [14].
Most patients with renal involvement show a poor response to treatment. Approximately half develop renal insufficiency and die within two years, on average [12]. The patient described here developed a new bone lesion within a period of two years; however, his renal lesion did not show progression. Although a follow-up period of two years is short, a wait-and-see policy can be adopted in ECD patients with an asymptomatic renal lesion.
 XML Download
XML Download