

 PDF
PDF Citation
Citation Print
Print


Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a distinctive skin lymphoma that is characterized by infiltration of neoplastic cytotoxic T-cell mimicking panniculitis [1,2]. SPTCL presents with multiple erythematous subcutaneous nodules, usually in the absence of other sites of disease. The most common sites of localization are the extremities and trunk. The nodules range in size from 0.5 cm to several centimeters. The condition is sometimes associated with fulminant hemophagocytic syndrome (HPS), characterized by fever, pancytopenia, hepatosplenomegaly, and coagulopathy [2-5]. The natural history of SPTCL is highly variable ranging from a chronic relapsing to a rapidly fatal course. However, if HPS is present, the prognosis is poor and sometimes fatal despite aggressive chemotherapy [2-5].
Treatment strategies for SPTCL with HPS remain controversial because of the rarity of this condition. Anthracycline-based regimens, cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) or CHOP-like combinations have been used most frequently for patients with aggressive disease, but the overall outcome remains poor [3]. Although there are several case reports that show high dose chemotherapy followed by hematopoietic stem cell transplantation (HSCT) to be effective in patients with chemotherapy refractory SPTCL, the role of HSCT has not been fully established [3-5]. We report a 35-year-old woman with SPTCL complicating HPS diagnosed by histopathology and immunohistochemistry. This case showed only transient responses to combination chemotherapy and autologous HSCT, but displays a sustained complete remission after a course of cyclosporine with prednisolone.
Go to : 
Case Report
A previously healthy 35-year-old woman presented with intermittent high fever, chills, and fatigue, as well as multiple and painful subcutaneous nodules on the lower abdomen for 3 months. Despite treatment with antibiotics and non-steroidal anti-inflammatory drugs, she continued to experience a spiking fever of 39℃, and the tender nodule increased. Her medical and family histories were unremarkable. On physical examination, the patient was pale and febrile. There was moderate splenomegaly but no lymph node enlargement and hepatomegaly. Skin examination revealed multiple erythematous, tender, and firm subcutaneous plaques of variable size (12 × 9 cm, 5 × 3 cm) on the lower abdomen. The laboratory investigation included the following: white blood cell count of 1,800/µL, hemoglobin level of 10.0 g/dL, platelet count of 124,000/µL, erythrocyte sedimentation rate of 48 mm/hr, C-reactive protein of 32.5 mg/L, elevated transaminase (aspartate aminotransferase of 112 IU/L, alanine aminotransferase of 51 IU/L), high low-density lipoprotein (1,878 IU/L), high ferritin (3,249 µg/L), triglyceride of 139 mg/dL, and D-dimer of 2.27 ug/mL. Throat, midstream urine, sputum, and blood culture results were negative. Also, serologic test for syphilis, human immunodeficiency virus, and hepatitis B and C viruses were negative. However, Ebstine-Barr virus and cytomegalovirus serologic values revealed evidence of past infection. Tuberculin and Coombs test were negative. Rheumatoid factor and anti nuclear antibody were negative as well. Abdominal computed tomography revealed multiple ill-defined enhancing subcutaneous nodules in the abdominal wall (Fig. 1A). Whole body 18F-fludeoxyglucose-positron emission tomography scan image showed multiple hypermetabolic soft tissue nodules in the subcutaneous layer all over the body (Fig. 1B). Bone marrow examination showed increased histiocytes and evidence of hemophagocytosis. An excisional biopsy of the nodule from the lower abdomen showed the rimming of the lymphoid cells around individual fat cells. The overlying dermis and epidermis were not involved. Immunohistochemistry analysis showed that tumor cells expressed CD3 and CD8 but were negative for CD56 (Fig. 2). T-cell receptor (TCR) (γ chain) gene rearrangement was negative following polymerase chain reaction analysis. Although the status of T-cell gene rearrangement was not confirmed, rimming of individual adipocytes by the CD8+ and CD56- neoplastic T-cell and strict subcutaneous localization were consistent with a diagnosis of SPTCL.
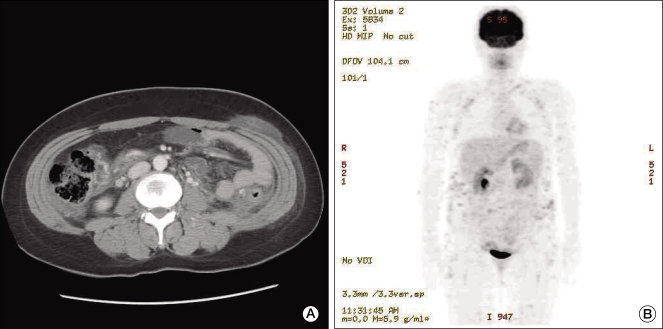 | Fig. 1Radiologic finding. (A) Abdominal computed tomography shows an enhancing nodule in the subcutaneous layer of the low abdominal wall. (B) Positron emission tomography shows numerous fludeoxyglucose uptakes in the subcutaneous layer of the whole body.
|
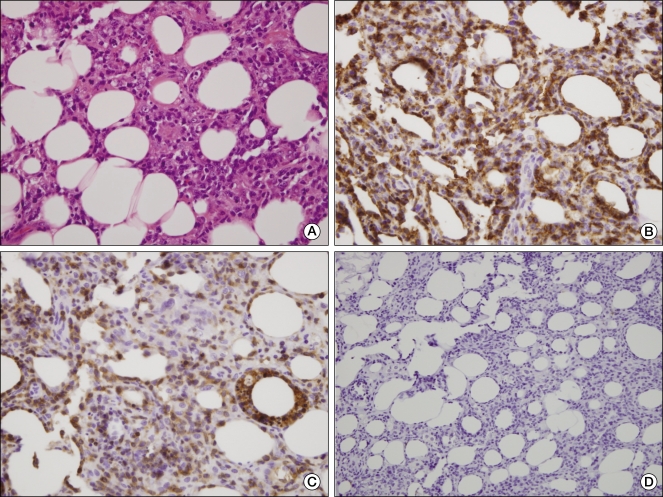 | Fig. 2Tumor pathology. (A) Histopathology of subcutaneous nodule showing neoplastic lymphoid cells infiltrating mainly lobular areas of subcutaneous tissue (H&E staining, ×400). Immunohistochemical stains showing the rimming of individual fat spaces by tumor cells with staining for CD3 (B) and CD8 (C) (H&E staining, ×400). (D) Tumor cells do not express the CD56 phenotype (H&E staining, ×200).
|
The patient was treated with CHOP regimen, resulting in the transient disappearance of the plaques and fever. Two months after completing 6 cycles of chemotherapy, her disease relapsed with multiple nodules on both arms. She received 4 additional cycles of ifosfamide, carboplatin, etoposide regimen. After salvage chemotherapy, complete remission was achieved, and autologous HSCT was undertaken with a conditioning regimen comprising busulfan, cyclophophamide, and etoposide. Her neutrophil and platelet count recovered within 3 weeks and no severe major organ impairment occurred during HSCT. Four months after the autologous HSCT, a new lesion on the left breast was confirmed by clinical examination and biopsy. The patient underwent 2 additional cycles of chemotherapy and at the end of these cycles returned with multiple erythematous subcutaneous nodules, febrile neutropenia and severe bone marrow suppression. Therefore, chemotherapy was discontinued. Eight months after the autologous HSCT, the patient was started on cyclosporine 4 mg/kg/day and prednisone. The fever and other systemic symptoms and skin lesions resolved within 2 months after the treatment. Prednisone administration was tapered gradually, and a maintenance dose of cyclosporine was continued. The patient's condition remained in complete remission at 20-month follow-up. There was no evidence of clinical relapse.
Go to :
DISCUSSION
Gonzalez et al. [1] first described SPTCL in 1991. It is a rare form of lymphoma, representing less than 1% of all non-Hodgkin lymphomas. SPTCL is slightly more common in females than males and has a broad age range. Some patients present with HPS and the expression of CD56 by tumor cells, both of which are associated with shorter survival [3,4]. It also had been observed that SPTCL's clinical course was different depending on the TCR phenotype and immunophenotypic characteristics of the tumor cells. Therefore, a distinction had been made between cases with a α/β T-cell phenotype and with a γ/δ T-cell phenotype, with the latter typically expressing the CD56 phenotype and being more aggressive [3-6]. In the fourth edition of the World Health Organization (WHO) classification, cases expressing the γ/δ TCR are excluded from SPTCL, and reclassified as primary cutaneous γ/δ T-cell lymphoma [2]. Although TCR rearrangement analysis and immunophenotyping with TCR βF1 were not available in our patient, rimming of individual adipocytes by the CD8+ and CD56- neoplastic T-cell and strict subcutaneous localization were consistent with a diagnosis of SPTCL.
The best treatment strategy for SPTCL has not been fully defined because of its rarity and clinical diversity. In general, immunosuppressive agents, such as prednisone and cyclosporine, or low-dose chemotherapy with a single agent were used as the initial treatment for patients who had a relatively indolent presentation. For patients with a more aggressive disease, up-front anthracycline-based combination chemotherapy was used most frequently [3-5]. Because the presence of HPS at diagnosis is one of the most important prognostic factors that predict poor overall survival, it may be a parameter that determines whether disease is indolent or aggressive [3,4]. The other factors associated with an unfavorable prognosis are low white blood cell count or elevated lactate dehydrogenase (LDH) [5]. Our patient presented multiple subcutaneous lesions, accompanied with HPS, and had elevated LDH at the time of diagnosis. Her condition was diagnosed as an aggressive form of SPTCL and she was treated with CHOP as the first line of treatment.
There have been conflicting results about the role of up-front anthracycline based combination chemotherapy. An analysis of patients with SPTCL in the literature reported that CHOP or CHOP-like therapies resulted in an overall response of 53% and median duration of complete remission of more than 11.5 months [3]. In addition, a case report showed durable remission in SPTCL with HPS after CHOP [7]. However, the report from a workshop of the European Organisation for Research and Treatment of Cancer Cutaneous Lymphoma Group showed only two of 11 SPTCL patients with HPS exhibited durable survival after CHOP(-like) therapies [4]. These results indicate that anthracycline based chemotherapy is not very successful in patients with SPTCL with associated HPS. In this case, although she achieved complete remission during CHOP therapy, the duration of remission was short. Anthracycline based combination chemotherapy as first line treatment in SPTCL patients with HPS should be questioned.
Autologous HSCT is the standard consolidation treatment after salvage chemotherapy in a relapsed lymphoma, provided the lymphoma has been chemosensitive to the conventional salvage chemotherapy regimen. However, the role of autologous HSCT in the SPTCL has not been clarified because most information comes from case series. Some patients with SPTCL have recently undergone high dose chemotherapy followed by autologous HSCT, and most of them achieved complete remission with a median follow-up of 14 months [4,5,8-10], but the possibility that failure after HSCT may be under-reported can not be ruled out. In the present case, the patient only had transient remission after autologous HSCT, even though she had a complete remission during salvage chemotherapy.
Our patient showed only transient responses to CHOP and high-dose chemotherapy, but displayed a marked complete remission after cyclosporine and prednisolone without subsequent chemotherapy. Cyclosporine had been suggested as a potentially effective agent in the treatment of angioimmunoblastic T-cell lymphoma, a disease with considerable immune dysregulation [11]. Several reports suggest that patients with SPTCL without HPS may benefit from variable immunosuppressive agents including cyclosporine and steroid [4,12,13]. Serum levels of interferon γ and soluble interleukin-2 receptor were elevated during the active disease and normalized after cyclosporine, suggesting that cytokine upregulation and modulation may be the pathophysiology of SPTCL as well as the mechanism of action of this drug [13]. However, to our knowledge, this is the first report which shows the efficacy of cyclosporine in relapsed SPTCL with HPS after HSCT. This result suggests that patients with SPTCL may benefit from immunosuppressive therapy even though it is a clinically aggressive disease. Further study on novel therapeutic strategies, including cyclosporine containing immunosuppression as a first line of treatment, is needed to improve the clinical outcome of SPTCL with HPS.
Go to :
 XML Download
XML Download