

 PDF
PDF Citation
Citation Print
Print


Introduction
Chronic myelogenous leukemia (CML) is a clonal myeloproliferative disorder characterized by the BCR-ABL tyrosine kinase, reciprocal t(9;22) (q34;q11) chromosomal translocation (1). Targeted inhibition of this active kinase with imatinib is highly effective and is a standard therapy for patients with CML (2). Despite the promising high rates of hematologic and cytogenetic responses, resistance has been reported in a population of patients receiving imatinib therapy (3). The various mechanisms underlying this resistance are heterogeneous and one of the well-defined mechanisms is the expansion of leukemia subclones with mutations in the BCR-ABL kinase domain (4,5). Dasatinib and nilotinib inhibit the activity of most mutations in patients with imatinib resistance, except for T315I, and have significant activity in patients with CML after imatinib failure (6-8). However, subtle differences exist between in vitro observations and clinical entities in a small subset of mutant clones (9,10). Here, we identified three patients who showed resistance to a second generation tyrosine kinase inhibitor (TKI), as compared to in vitro observation, and developed chromosomal abnormalities.
Go to : 
Materials and Methods
1. Patients
We quantified various mutated BCR-ABL transcripts during the follow-up of eight patients treated with dasatinib or nilotinib. The patients were entered into the BMS-354825 clinical trials (studies CA180-005, 180-034, 180-109, and 180-188) or AMN107 clinical trial (studies CAMN-107A2101 or 107A2109) between June 2005 and April 2009. The physicians explained the nature of the study to the patients and informed consent was obtained. The institutional review board of Chonnam National University Hwasun Hospital, Republic of Korea, approved this study. The median follow-up was 23 months (range, 3~41 months).
2. Quantitation of BCR-ABL mRNA
Total RNA from bone marrow aspirates was extracted with a total RNA isolation kit (LeukoLOCK™; Ambion, Austin, TX). The cDNA was synthesized using random hexamers with a Superscript reverse transcriptase kit (Invitrogen, Carlsbad, CA). All BCR-ABL fusion transcripts were examined using the Rotor-Gene 3000 system (Corbett Research, Australia) and TaqMan probes (BCR-ABL commercial kit; Biosewoom, Korea). The BCR-ABL transcripts were quantitatively expressed as the BCR-ABL/ABL ratio.
3. DNA pyrosequencing
When BC-ABL mutations were detected in a patient by direct DNA sequencing, the samples were subjected to DNA pyrosequencing in accordance with the manufacturer's instructions (Pyrosequencing, Uppsala, Sweden). The BCR-ABL amplicon was used as a template for nested PCR with primers flanking the region containing the putative mutation. PCR primers and sequencing primers were designed using the Pyrosequencing SNP Primer Design Software (version v1.0.6). The forward primer was biotin-labeled and the PCR products were purified with streptavidin Sepharose HP beads (Amersham Biosciences, Piscataway, NJ). The pyrophosphate released by incorporating a complementary nucleotide into the sequencing primer was converted to adenosine 5' triphosphate (ATP) by sulfurylase. The ATP generated was utilized as a coenzyme for luciferase that oxidizes luciferin to oxyluciferin, thereby emitting light. The light intensity was proportional to the level of the single-nucleotide polymorphism in any given sample. The percentages of the mutant and non-mutated alleles were determined by the allele Quantitation Algorithm using the PSQ 96MA system (Pyrosequencing).
4. Cytogenetic analysis
Conventional cytogenetic analysis was performed on G-banded preparations from 48 h unstimulated cultures of peripheral blood cells. The chromosome aberrations were described according to the International System for Cytogenetic Nomenclature 2005.
Go to :
Results
We monitored the kinetics and interrelationship of the clones expressing the non-mutated and mutant transcripts in eight patients with CML receiving a second TKI. Details of the mutations and cytogenetics prior to treatment and during the second TKI treatment are shown in Table 1. Of the eight patients evaluated, four were in the chronic phase, three in the accelerated phase, and one in blast crisis at the start of the second TKI administration. Five patients were still taking dasatinib; the drug had been discontinued in three patients because of disease progression. Patients were classified into two patterns based on the correlation with in vitro drug sensitivity data based on the pattern of changes over time and the mutant BCR-ABL transcripts. Examples of representative patterns are shown in Fig. 1. The total level of BCR-ABL transcripts did not initially decrease during imatinib treatment; however, the level of BCR-ABL transcripts and the percentage of mutant alleles decreased after changing to the second TKI (P#1: H396R; P#2: F359I; P#3: G250E). No clinical response was observed in patient #4 (T315I) after 9 months, and the patient died of disease progression. The response of four patients (P#1, P#2, P#3, and P#4) correlated with in vitro drug sensitivity to dasatinib. Patients #5, #6, #7, and #8 (#5 and #7, E255K; #6; G250E; #8, M351T) showed resistance to the second TKI compared to in vitro drug sensitivity. The original mutant clone disappeared in patient #8 (M351T) and this correlated with a decrease in BCR-ABL transcripts. This patient lost the original mutations but acquired a new mutation (H396R), which caused an increase in the number of BCR-ABL transcripts. Patients #5, #6, and #7 had no additional mutant clones. These patients developed complex chromosomal abnormalities during the second TKI therapy. Patients #5 and #7 showed new additional chromosomal evolution in Philadelphia (Ph) chromosome positive metaphases at 12 and 2 months, respectively, after administration of the second TKI. 6 months after beginning the second TKI, clonal evolution occurred in Ph negative cells of patient #6.
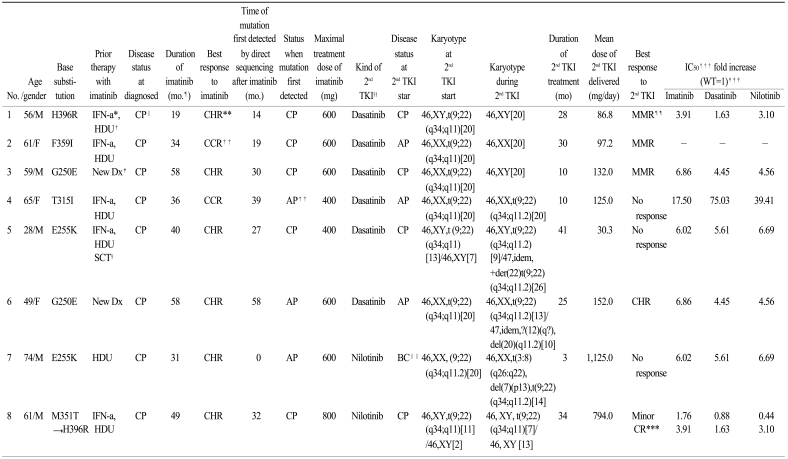
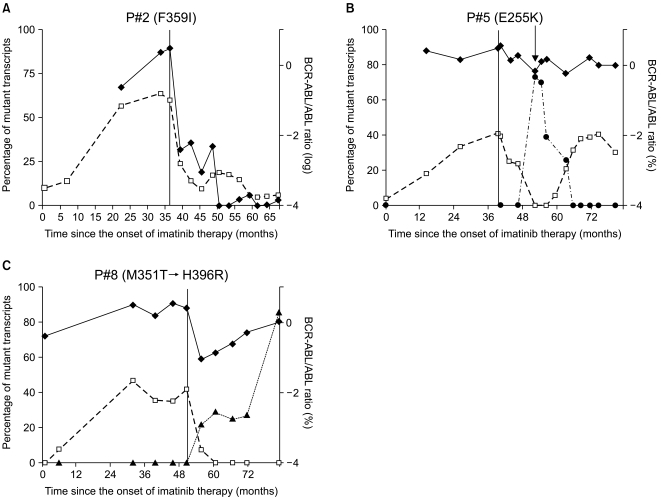 | Fig. 1Kinetics of all BCR-ABL transcripts and the percentage of mutant transcripts in representative patterns. Longitudinal line in graphes represent the time point of treatment with second TKIs and diamonds (◆) depict the quantitation of BCR-ABL transcripts by RQ-PCR expressed as BCR-ABL/ABL ratios on a log scale. Squares (□) and triangles (▲) represent the proportion of BCR-ABL mutated transcripts expressed in the percentage of total BCR-ABL transcripts. Round (●) symbols represent the percentage of chromosomal abnormalities in the Philadelphia chromosome. The arrow indicates the time point for a developed clonal evolution. The response of patient #2 (F359I) was correlated with in vitro sensitivity to dasatinib (A). Patient #5 (E255K) showed resistance to the second TKIs as compared to in vitro observation and developed clonal evolution during dasatinib treatment (B). Patient #8 (M351T → H396R) lost his original mutations but acquired a new mutation (H396R), which was the cause of the increase in BCR-ABL transcripts (C).
|
Table 1
Details on patients with detected kinase domain mutations
*interferon-a, †hydroxyurea, ‡new diagnosis, §stem cell transplantation, ∥chronic phase, ¶month, **complete hematologic response, ††complete cytogenetic response, ‡‡accelerated phase, §§second generation tyrosine kinase inhibitor, ∥∥blast crisis, ¶¶major molecular response, ***cytogenetic response, †††relative concentration that inhibit 50% cell viability; IC50 values are from Redaelli et al. 2009 (8), ‡‡‡wild type.
![]()
Go to :
Discussion
Dasatinib, a dual Src and Abl-kinase inhibitor, is approximately 300-fold more potent than imatinib and nilotinib, which have no activity against Src kinases. Nilotinib is 30-times more potent in vitro against imatinib-sensitive BCR-ABL expressing cells (7,11). BCR-ABL kinase is reactivated in patients who relapse on imatinib therapy, emphasizing the importance of the kinase activity of this protein to disease pathogenesis. The most common mechanism of resistance involves specific mutations in the BCR-ABL kinase domain that interfere with imatinib binding without eliminating ATP binding or kinase activity (4). Dasatinib and nilotinib inhibit the activity of most mutations observed in patients with imatinib resistance, except for T315I, and they have significant activity in patients with CML after imatinib failure (6-8). However, subtle differences between in vitro observations and clinical entities have been observed in some mutant clones (10).
Various response levels with the use of dasatinib has been reported (10). Based on the BCR-ABL mutation phenotype, the researchers observed different levels of response in vivo, suggesting other that mechanisms were responsible for the lack of complete sensitivity to dasatinib. Another study reported that some mutant subclones (G250R and M244V) did not achieve major molecular response despite continuing dasatinib, and dasatinib resistance was not attributable to the mutant subclone but to other undefined mechanisms (12). Some studies have reported the development of new mutant clones after the disappearance of the original mutant clone. Sequential, newly detected mutant clones result from a selected advantage during the second TKI treatment (12-14). As patients are treated with a TKI, the balance of clones may change, and sensitive clones may be suppressed and more resistant clones may emerge (13). The resistance to the second TKI in patient #8 can be explained by the appearance of a sequential-resistant mutant clone, whereas explaining the resistance to the second TKI in patients #5, #6, and #7 is difficult.
Cytogenetic clonal evolutions in patients with CML on imatinib therapy show poor prognostic significance for survival, perhaps because of its relative independence from the BCR-ABL-related molecular events (15). Alternative, partially or totally independent mechanisms are presumed to drive the growth and survival of the malignant clone in this form of imatinib resistance (16). In the dasatinib era, a similar result was reported (17). Patients with new chromosomal evolutions in Ph positive metaphases did not achieve a cytogenetic response. We could explain the mechanism of resistance in patients #5 and #7.
Clonal evolutions in Ph chromosome negative cells during imatinib treatment of Ph positive CML have been observed (18-20). Jabbour et al. (18) reported that patients with clonal evolutions in Ph chromosome-negative cells had a significantly inferior prognosis for overall and progression-free survival. These findings reflect genomic instability of the hematopoietic cells of patients with CML. CML pathogenesis has been proposed in a multistep model (19). Tyrosine kinase could select these preexisting chromosomal changes (19,21). This suggests that the Ph translocation induces CML with the contribution of another clonal change. Other studies have reported that clonal evolutions in Ph chromosome-negative cells do not impair the cytogenetic response to tyrosine kinase (17,20). In our study, patient #6 developed a clonal evolution with a BCR-ABL mutant in Ph negative cells. These complex mechanisms easily allow compromising genomic surveillance mechanisms that might characterize the advanced stages of CML.
Go to :
 XML Download
XML Download